
Quality management for medical devices refers to the systems and processes put in place to ensure that medical devices are safe, effective, and meet regulatory requirements.
The ISO 13485 standard includes requirements for managing the entire lifecycle of a medical device, from design and development to manufacturing, distribution, and post-market surveillance.
This illustrated guide was written by Peter Sebelius, a member of the Joint Working Group who authored the latest versions of ISO 13485 and ISO 14971.
This guide will:
- Provide a useful overview of quality management and ISO 13485,
- Clarify terminology used in the standard that is defined elsewhere, and
- Address common misconceptions.
Navigate quality management essentials in our illustrated guide, now as an ebook!

Why is quality management important?
Quality management is not only important for selling products that meet or exceed customer requirements. In the medical device industry, quality management is also important for regulatory compliance and market access.
Medical device organisations must comply with various regulations to ensure their devices meet certain quality requirements. The ISO 13485 standard is important, if not the most important standard, and can be used to fulfil the requirements of the regulations.
What is quality management?
When definitions of key terms are missing in ISO 13485, one should not primarily turn to dictionaries but rather read ISO 9000:2015 Quality management systems – Fundamentals and Vocabulary.
In ISO 9000:2015, quality management is defined as “management with regard to quality”. This definition is not particularly useful, but if the terms from the definition are expanded, it makes more sense.

Quality management can be seen as coordinating activities to direct and control an organisation with regard to fulfilling requirements on a product, service, process, or system.
What is a quality management system (QMS)?
Working successfully with quality management is virtually impossible without a Quality Management System, or QMS.
A QMS is a set of policies, processes, and procedures implemented by an organisation with the main objective of managing quality.
Paper-based vs electronic quality management systems
Quality management systems can either be paper-based or electronic. Each type comes with its own unique set of advantages and challenges.
A paper-based system would typically be a number of documents found in binders or scanned versions of signed paper copies that can be accessed through an internal network.
A paper-based QMS is easy to implement and does not require much of an initial investment, but as an organisation grows, paper-based systems are more prone to human error and slower in their operations.
An electronic QMS, or eQMS, typically contains procedures and templates as electronic documents approved and distributed completely electronically.
An eQMS is often faster, more efficient, and can reduce the risk of errors, but the implementation may require a significant investment.
A hybrid QMS uses the best of the two worlds of paper-based QMS and eQMS and uses a mix of paper-based documents and electronic documents.
Quality management requirements for medical devices in the EU
All medical devices placed on the European Union market must meet the regulatory requirements of the EU. This is mandated by the Medical Device Regulation (MDR) and the In-Vitro Diagnostic Medical Device Regulation (IVDR).
Section 9 in Article 10 of the MDR/IVDR clearly stipulates that organisations of devices shall establish, document, implement, maintain, keep up to date and continually improve a quality management system.
The QMS is integral in ensuring that the development, manufacturing and other operational processes fulfil quality and safety requirements, thereby producing medical devices that are safe, effective and regulatory compliant.
ISO 13485: A detailed guide to quality management systems
While the MDR and IVDR provide a high-level quality management requirements, ISO 13485 delves much deeper into the details. The ISO 13485 standard is 70 pages long, in contrast to the MDR, which all in all is about 170 pages, but only includes about one page with requirements specifically on the quality management system.
ISO 13485 is harmonised with the MDR and IVDR. The harmonisation of ISO 13485 means that if the requirements of the ISO 13485 are met, it can be presumed that the corresponding requirements of the MDR and IVDR are also met.

While conforming to harmonised standards, such as ISO 13485, is not explicitly obligatory, it is decidedly desirable. It is important to note that any deviation from ISO 13485 or other applicable harmonised standards necessitates robust justification.
More information on where you can download the ISO 13485 standard here.
Quality management requirements for medical devices in the US
EU has two different regulations for medical devices (MDR) and in-vitro diagnostic medical devices (IVDR) respectively, in the United States, both medical devices and in-vitro diagnostic medical devices are subject to the same regulation; the 21 CFR 820 or Quality System Regulation (QSR).
The Food and Drug Administration (FDA) is actively working towards aligning the Quality System Regulation with ISO 13485. The resulting regulation, once realised, will be referred to as the Quality Management System Regulation (QMSR). You can find the latest news on the QMSR here.
The equivalent to harmonised standards in the US context is recognized consensus standards. ISO 13485 is not a recognised consensus standard for the US market as the QSR includes requirements in the same areas as the ISO 13485.
ISO 13485 requirements on quality management systems
The QMS must be documented and implemented in a way that is appropriate for the size and complexity of the organisation and the types of medical devices manufactured. The ISO 13485 standard can be applied to anything from a one person company to a multinational company with thousands of employees.
The ISO 13485 can also be applied to companies with very different types of operations; it could be a consulting company, or a manufacturer of complex medical devices. This means that there is not just one acceptable solution or one size fits all QMS that will satisfy the requirements of the ISO 13485. On the contrary, there is an infinite number of ways to implement the requirements of ISO 13485.
The QMS should not be a static set of documents. It must be regularly reviewed and updated to ensure its continued effectiveness.
Organisations are required to establish a quality manual. The quality manual is the top-level document for the QMS and the starting point for accessing the Quality Management System (QMS).
The quality manual must outline the structure of the documentation used in the quality management system. An overview, similar to the one presented below but without the explanatory texts, is commonly used to fulfil the requirement.

Furthermore, ISO 13485 requires 31 documented procedures. These documented procedures are the pillars of your Quality Management System (QMS). The 31 documented procedures do not necessarily have to be 31 physical documents, but a documented procedure can be split up and found in several documents. Or several documented procedures can be merged into one physical document.
The required documented procedures are typically found in Standard Operating Procedures or short SOP. SOPs serve as the organisation’s manuals, instructions, and training material, instructing employees on how to carry out work.
Documented procedures required by ISO 13485
|
4.1.6 Validation of the application of computer software used in the quality management system
|
7.5.11 Preserving conformity of product to requirements during processing, storage, handling, and distribution
|
|
4.2.4 Controls needed to review, approve documents, update and re-approve documents
|
7.6 Procedures to ensure monitoring and measurement is done according to requirements
|
|
4.2.5 Controls needed for the identification, storage, security and integrity, retrieval, retention time and disposition of records
|
7.6 Validation of computer software used for monitoring and measurement
|
|
5.6.1 Management review
|
7.6 Calibration and verification
|
|
6.4.1 Monitor and control work environment if it has effect on product quality
|
8.2.1 Feedback
|
|
7.3.1 Design and development
|
8.2.2 Complaints handling
|
|
7.3.8 Transfer of design and development outputs to manufacturing
|
8.2.3 Notification of adverse events or issuance of advisory notices
|
|
7.3.9 Control of design and development changes
|
8.2.4 Responsibilities and requirements for planning and conduct of audits
|
|
7.4.1 Ensuring purchased product conforms to purchasing information
|
8.2.6 Monitoring and measuring characteristics of the product
|
|
7.5.1 Procedures for control of production
|
8.3.1 Responsibilities and authorities for identification, documentation, segregation, evaluation, and disposition of nonconforming product
|
|
7.5.6 Validation of processes
|
8.3.3 Issuing advisory notices
|
|
7.5.6 Validation of computer software used in production and service
|
8.3.4 Rework
|
|
7.5.7 Validation of processes for sterilization and sterile barrier systems
|
8.4 Collect and analyze appropriate data
|
|
7.5.8 Identification of production by suitable means through product realization
|
8.5.2 Reviewing nonconformities
|
|
7.5.8 Returned medical devices
|
8.5.3 Determining nonconformities and their causes
|
|
7.5.9.1 Traceability
|
It is also recommended but not required to include templates and forms in the QMS.
Common examples of templates and forms that are useful to have include:
- Risk management plan template
- Hazard traceability matrix template
- Risk management report template
- Post-market surveillance plan template
- GSPR checklist template
- Complaints form
- CAPA form
- Change request form
- Change order form
- Concession form
- Product release form
The ISO 13485 also requires the organisation to establish, implement, and maintain any requirement, procedure, activity or arrangement required to be documented by applicable regulatory requirements; thus, the 31 documented procedures listed above may not be sufficient if the QMS should be in accordance with the requirement of the MDR or IVDR.
ISO 13485 requirements on having a quality policy
ISO 13485 requires that the top management of an organisation establishes and maintains a quality policy. The quality policy should be a statement that outlines the organisation’s commitment to meeting customer and regulatory requirements. It should also demonstrate the organisation’s commitment to maintaining the effectiveness of its quality management system (QMS).
Oftentimes quality policies contain information about continually improving the QMS. This is a spill over from requirements from the ISO 9000 series of standards that applies the concept of continuous improvements. The ISO 13485, being a regulatory standard, does not include a requirement to continuously improve, but only to reach the level required. Going beyond the level required may be desirable from a business point-of-view but is not a regulatory requirement in ISO 13485.
The requirement to continuously improve the QMS may still apply if the organisation aims to meet MDR requirements, because Article 10, section 9 of the MDR requires that:
"Manufacturers of devices, other than investigational devices, shall...continually improve a quality management system".
Below is an example of a generic quality policy for reference.

The quality policy must be consistent with the organisation’s overall business objectives and provide a framework for setting quality objectives. Quality objectives should be measurable and consistent with the organisation’s quality policy.
Examples of quality objectives according to ISO 13485 could be:
- The median time to answer support calls should be less than 5 minutes.
- The organisation should have less than four non-conformities from notified body audits every year.
Please note that quality objectives can sometimes be milestone-based and not a continuous measurement. This is useful to know when working with quality objectives for start-ups that don’t have continuous operations with a lot of data points. An example of such a milestone-based quality objective can be:
- The Xmed product shall be CE-marked before the end of 2025.
Effectiveness vs efficiency
The term “effective” is used both in conjunction with requirements on the quality policy as well as the quality objectives. ISO 13485 often refers to “effective” but never to “efficient”. The words “effective” and “efficient” both refer to achieving a result but with one important difference.
“Effectiveness” measures the extent to which planned activities are realised and planned results are achieved, and “efficiency” will add a dimension to this by including how many resources were spent to achieve the result.
From a regulatory point-of-view, it is never a requirement to be efficient; it is only a requirement to be effective. However, from a business point-of-view, it may be important to be efficient.
Who is top management in a medical device organisation?
Top management is defined as “a person or group of people who directs and controls an organisation at the highest level” in ISO 9000:2015. ISO 13485 states that top management is responsible for establishing and maintaining the Quality Management System (QMS) and ensuring that it is effective in meeting customer and regulatory requirements.
ISO 13485 requires organisations to establish a documented procedure for management review. The management review is a critical component of the quality management system as it provides top management with an opportunity to evaluate the performance of the system, identify areas for improvement, and make decisions on resource allocation to ensure that the QMS is aligned with the organisation’s strategic objectives and regulatory requirements.
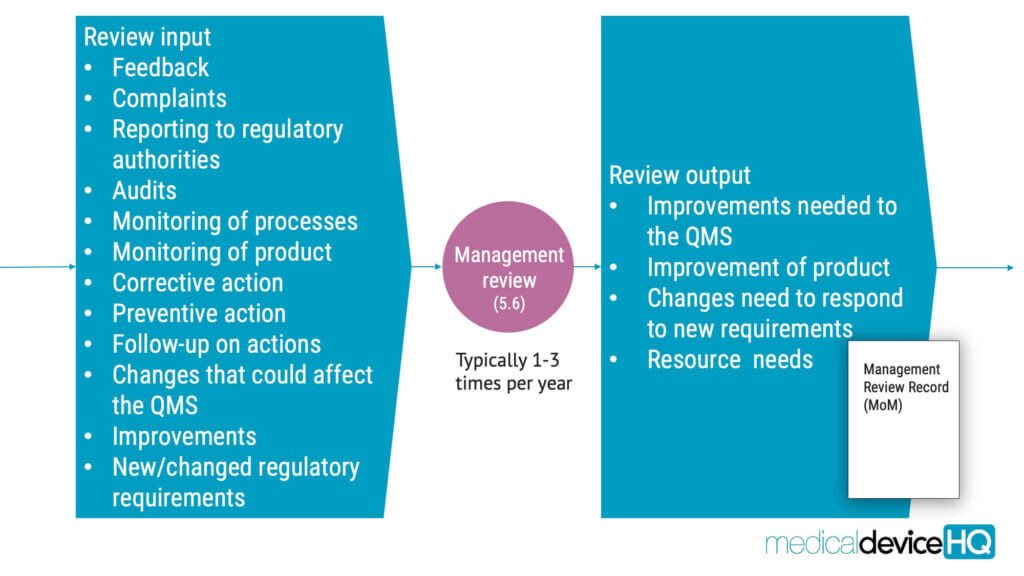
Medical device audits
One of the key components of quality management is to conduct audits to ensure that the QMS is effective.
Broadly, three distinct categories of audits exist, each with its unique significance and purpose.
First-party audits, also known as internal audits, are performed to determine the continued suitability, adequacy, and effectiveness of the quality management system and to identify improvement opportunities. This is the type of audit that is required by ISO 13485.
Second-party audits, or external audits, are performed by external providers to obtain and maintain confidence in the capability of an external supplier. This type of audit is not always explicitly required by ISO 13485, but it is very common to use second-party audits for supplier evaluation purposes. Not doing so should be seen as an exception to the rule.
Finally, there are two types of third-party audits: certification and/or accreditation and statuary or regulatory audits. The purpose of these third-party audits is to conform to applicable statutory and regulatory requirements. This category would include audits by notified bodies and the FDA.

ISO 13485 requirements on internal audits
Internal audits of a quality management system should be conducted at planned intervals to ensure that the system is effectively implemented and maintained. The ISO 13485 will not provide a lot of detail on how to carry out audits, but ISO 19011:2018 will. Find the latest version of the ISO 19011 standard here.
The purpose of the internal audit is to identify any nonconformities or areas for improvement within the quality management system.
The internal audit process should follow a documented procedure and include a schedule of planned audits, criteria for selecting auditors, and procedures for conducting the audit.
The ISO 13485 does not say much about the schedule, but the high-level schedule is referred to as audit program in ISO 19011.
The audit program is a set of one or more audits planned for a specific time frame and directed towards a specific purpose. For each audit, audit plans should be established. An audit plan includes a description of the activities and arrangements for an audit.
The term audit plan is often used incorrectly to refer to audit program. The overview below shows the relationship between an audit program and audit plans.

The audit must be conducted by trained personnel independent of the area being audited. The auditor or audit team should review relevant documentation, interview personnel, and observe processes to determine if the quality management system is meeting the requirements of ISO 13485 and other applicable audit criteria.
Following the audit, corrective actions should be implemented as needed, along with follow-up activities and verification results. Records of any audits performed must be maintained.
ISO 13485 requirements on risk management
The ISO 13485 standard requires organisations to “apply a risk-based approach to the control of the appropriate processes needed for the quality management system“. In this context, one must consider both risk as it relates to harm to people, property, and the environment, as it is required by ISO 14971, but also the risk of regulatory nonconformities.
Furthermore, ISO 13485 requires the organisation to document one or more processes for risk management in product realisation, where records of risk management activities carried out as part of the risk management process shall be maintained.
ISO 13485 refers to ISO 14971 for further information on risk management. This risk management standard is harmonised with the MDR and IVDR and is a recognised consensus standard for the US market.
Work with risk management relating to the safety of the medical device should be carried out in accordance with ISO 14971 requirements. This risk management process must identify, analyse, evaluate, and control all hazards and risks associated with the device throughout its entire lifecycle, from initial conception and design to production, distribution, use, and decommissioning.

ISO 13485 requirements on infrastructure and work environment
ISO 13485 requires that medical device organisations establish and maintain an infrastructure that is suitable for the production of safe and effective medical devices. This includes the physical environment, such as the building and facilities, as well as the equipment and tools used in the manufacturing process.

The work environment must be designed to prevent mix-ups and contamination and ensure that the medical devices are produced in a controlled and clean environment. This includes measures such as air filtration systems, temperature and humidity control, and regular cleaning and maintenance of equipment.
Documenting requirements on infrastructure and work environment
In the domain of infrastructure and work environment, documentation must be maintained. This includes not only the precise requirements for infrastructure but also any records related to the upkeep of such infrastructure.
Moreover, the health, cleanliness, and attire stipulations must be carefully documented. These factors play a pivotal role in maintaining quality and safety in the manufacture of medical devices.
It is also essential that staff members that work temporarily under special environmental conditions, such as in a clean room, are competent or supervised by a competent person.
Lastly, strict control of contamination and cleanliness as applicable during the production phase is critical. According to 6.4.2 in ISO 13485:
the organization shall document requirements for control of contamination with microorganisms or particulate matter and maintain the required cleanliness during assembly or packaging processes.
The above is commonly achieved by clean room production. Clean rooms are in turn covered by several standards, for example in the ISO 14644 series of standards.

ISO 13485 requirements to sell medical devices on the market
ISO 13485 requires that the organisation establish and maintain documented requirements for the product, including applicable regulatory requirements, requirements from the customer, user training, and additional requirements.
These requirements should be reviewed and updated throughout the product lifecycle to ensure that product requirements are defined and documented, regulatory requirements are met, and user training is available. Any changes must be documented, and records of reviews and actions must be kept.
ISO 13485 requirements on customer property
ISO 13485 requires medical device organisations to establish and maintain a system for the identification, documentation, verification, control, and safeguarding of customer property. This includes any property that is provided by the customer or external provider for use in the production or servicing of medical devices.
It is a common misconception that customer property only includes tangible goods. Intellectual property and personal data also qualify as customer property. See GDPR and HIPAA for more information relating to this area.
Examples of customer property include:
- Hardware
- Materials
- Components
- Intellectual property (IP) such as specifications, drawings, proprietary information
- Personal data
Communication with customers
Effective communication with customers is a crucial aspect of ISO 13485. The standard requires medical device organisations to establish and maintain communication processes with their customers to meet their needs and expectations.
This includes providing information about the device, its intended use, and any limitations or risks associated with it. This also involves documenting and investigating complaints, taking corrective actions, and keeping records of all customer complaints and feedback.
Communication with regulatory authorities
Communication with regulatory authorities is an essential part of ISO 13485 compliance. The standard requires medical device organisations to communicate with regulatory authorities in accordance with applicable regulatory requirements to ensure that they are aware of any changes in regulations or requirements that may affect their products.
ISO 13485 requirements on design and development
ISO 13485 requires medical device organisations to establish and maintain a documented design and development process.
This process must be appropriate for the specific type of device and involves a series of activities that starts from the initial concept and continues until the final product is released for distribution.
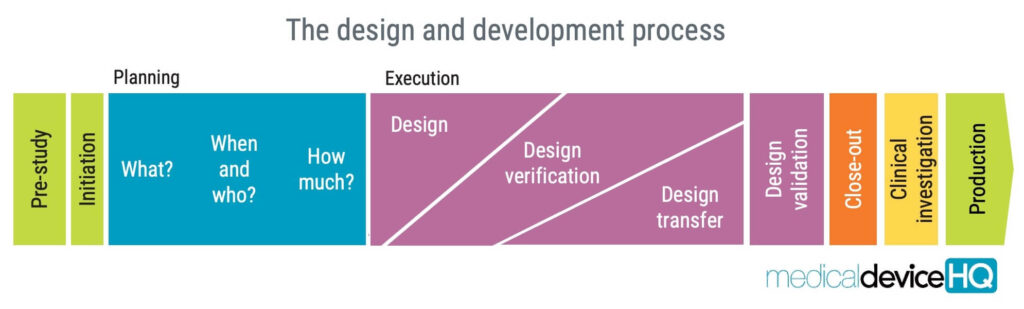
The requirements on design and development in ISO 13485 aim to ensure that the medical device meets the intended use, is safe and effective, and complies with regulatory requirements. From the manufacturer’s point-of-view, the process can also be used to achieve a product that customers will be happy with.
The design and development process involves several concepts, such as design and development planning, design and development inputs, design and development outputs, design and development review, design and development verification, design and development transfer, and design and development validation.
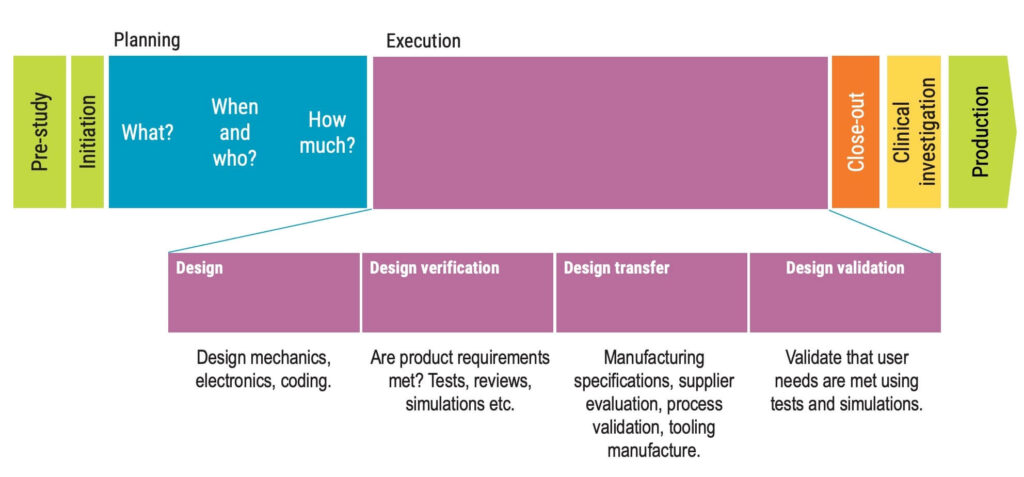
Each stage has specific requirements that should be determined by the organisation to ensure the quality of the medical device.
Usability and other regulatory requirements
Even though usability engineering is only mentioned a few times in ISO 13485, it is a critical component of risk management required by the FDA for the US market, and the MDR and IVDR for the EU market.
The ISO 13485 standard requires organisations to identify and assess the risks associated with the use of a device, including risks related to usability. Usability testing is an important tool for identifying and mitigating these risks.
By testing the device with representative users in realistic use scenarios, organisations can identify potential usability issues and make design changes to improve the device’s safety and effectiveness.
ISO 13485 requirements on product documentation
Product documentation should be maintained throughout the product lifecycle, from design and development through production, distribution, and post-market surveillance.
Organisations should also establish procedures for the retention of product documentation, including a design and development file, and medical device file. The QSR will refer to these as design history file (DHF), and device master record (DMR).
The MDR and IVDR will refer to “technical documentation” in Annex II and III, which is overlapping with the terms from ISO 13485.

ISO 13485 requirements on suppliers
Supplier evaluation is an important aspect of ISO 13485 and involves assessing the capability and performance of suppliers to ensure that they meet the requirements of the organisation. This may include that the suppliers should also meet ISO 13485 requirements.
ISO 13485 requirements on supplier evaluation
The evaluation process includes:
- Identifying and selecting suppliers,
- Establishing criteria for evaluation,
- Conducting the evaluation, and
- Monitoring supplier performance over time.
The criteria for evaluation must include factors such as ability to meet the organisation’s requirements and the supplier’s performance. From a regulatory point-of-view, price is usually not part of the criteria. The evaluation may be conducted through receiving prototypes, supplier surveys, audits, or on-site visits.
It is common for medical device organisations to establish an approved supplier list, but it is not explicitly required to have such a list, as there are other ways of keeping track of which suppliers are approved and not.
Organisations must have a plan in place to monitor and re-evaluate any suppliers. If a supplier delivers a product that is not according to specification, this is considered a nonconformity according to ISO 13485, 8.3.
It is required to maintain accurate records of supplier and purchase information. This also helps organisations to evaluate supplier performance. By tracking supplier performance metrics such as on-time delivery, product quality, and responsiveness to issues, organisations can make informed decisions about which suppliers to continue working with and which to replace.
ISO 13485 requirements on purchasing
ISO 13485 requires that medical device organisations establish and maintain procedures to ensure that the purchased products conform to specified purchasing information. These procedures should include requirements for the approval of purchased products or services, as well as the control of nonconforming products or services.
ISO 13485 also requires that medical device organisations maintain records of purchasing activities to fulfil requirements on traceability.
ISO 13485 requirements on production
Purchased goods and processed materials that are purchased for the production of medical devices must be preserved to ensure that the resulting products conform to requirements. This may include procedures relating to the storage and protection of the goods, materials and eventually finished products with appropriate packaging. Packaging should always be seen as an integral part of the design.
Medical devices that are delivered sterile to customers must be packaged in a sterile barrier system, and have protective packaging, designed to prevent damage to the sterile barrier system and its content from assembly to the point of use. The sterile barrier system must be evaluated from a usability point-of-view according to ISO 11607-1:2020. The acceptance criteria for such an evaluation are, however, not defined in ISO 11607-1.
Purchased products must be subject to incoming or receiving inspection. In ISO 13485, this is referred to as verification of purchased product. It is not explicitly mentioned that statistical methods should be applied when verifying purchased products, but it is most certainly expected to use sampling plans based on statistical rationales or that 100% of the purchased products are verified. Any products that are found to be nonconforming shall be processed according to procedures for nonconforming products.
Throughout the production process, the goods used in the production and the resulting products must be possible to identify. These requirements can be found in 7.5.8 of ISO 13485.
Practically speaking, identification can be done by, for example, placing goods in labelled boxes or putting stickers on them. Also, when applicable, it is required to document a system to assign UDI to the medical devices.
Appropriate controls must be applied throughout the production process to ensure that the products will conform to the requirements. The monitoring must be done according to plans. When measuring is carried out, the measuring instruments must be calibrated. Keep in mind that in-process controls in production will often be risk control measures; protective measures in the manufacturing process according to the ISO 14971 standard on risk management.
For every medical device or batch of medical devices manufactured, a batch record must be created. A commonly used term for batch record is Device History Record, or short DHR from the quality system regulation.
Product release, which is to allow the product to be placed on the market, shall be done according do documented arrangements. This typically means that it should be documented what tests, records, actions, reviews and approval of production records must be completed before releasing the product. For the EU market, the product release shall be carried out by the PRRC; Person Responsible for Regulatory Compliance.
If the medical device requires installation, the requirements of the installation must be documented.
Traceability requirements for medical devices
ISO 13485 does not, in itself, contain any details relating to traceability requirements other than that you must define procedures for traceability. For more details, ISO 13485 refers to applicable regulatory requirements for traceability.
As a general minimum, the manufacturer and its distributors must keep track of to whom and where the medical devices are delivered. This must be done to ensure that appropriate field safety corrective actions can be carried out should a need to do so arise.
More granular requirements for traceability apply to implantable devices according to 7.5.9.2
Manufacturers may decide to have more granular traceability for reasons of risk to patients and business risk. Having too little information about the medical devices delivered to customers can result in having to recall every medical device on the market instead of a particular batch or individual device.
ISO 13485 requirements on process validation
ISO 13485 requirements on process validation are an essential part of ensuring the safety and effectiveness of medical devices.
Process validation is the process of establishing evidence that a process is consistently producing a product that meets its predetermined specifications and quality attributes. This is achieved through a series of activities that include planning, design, qualification, and ongoing monitoring of the process.

Examples of medical device manufacturing processes that require process validation include injection moulding, printing, gluing, soldering, welding, and heat treating.
Other examples include the sterilisation process, assembly process, and packaging process. For instance, the sterilisation process must be validated to ensure that it effectively kills microorganisms on the device without damaging its functionality or causing any adverse effects on the patient.
ISO 13485 also requires software that is used as in the QMS, in production and service, and for monitoring and measurement of requirements to be validated. This means that if the organisation uses an eQMS, is must be validated.

Read more about process validation relating to production processes in the Global Harmonization Task Force document and about validation of software used in the QMS in ISO/TR 80002-2:2017 Medical device software – Part 2: Validation of software for medical device quality systems.
ISO 13485 requirements on nonconforming products
ISO 13485 requirements on nonconforming products are an essential aspect of the standard. Nonconforming products refer to products that do not meet the specified requirements.
The standard requires that organisations establish and maintain documented procedures for identifying, documenting, evaluating, segregating, and disposing of nonconforming products.
These procedures should include the identification, documentation, segregation, evaluation, and disposition of nonconforming products and the implementation of corrective actions to prevent recurrence of the nonconformity.
The standard also requires that the organisation review and analyse nonconforming products to identify trends and take appropriate action to address them.
Nonconformities detected before delivery of product
If a nonconforming product is detected before delivery, it is important to take action to prevent the product from being shipped to the customer.
The nonconforming product should be segregated and clearly marked to prevent it from being shipped or used. Nonconforming products are often marked with stickers and/or placed in a segregated area.
Depending on the criticality and the inherent risk of the nonconformity, the product may be:
- Eliminated
- Used under concession, or
- Reworked;
- Have parts replaced,
- Repaired, or
- Scrapped.
If rework is carried out, the organisation must verify that the product still meets the requirements and the intended use. A common question is whether a sterile medical device can be resterilised and then released to the market. Some manufacturers will pre-emptively perform a validation of a re-sterilisation process as a project risk control measure to prove that the product meets the requirements even after re-sterilisation.
Records shall always be kept for any rework or concessions.
Nonconformities detected after delivery of product
If a nonconforming product is discovered after it has been delivered to the customer, actions must be taken based on the effects of the nonconformity. This may include:
- Correcting the nonconformity
- Advisory notice;
- Sending out information on how to use the medical device safely, or
- Requesting the return or destruction of the medical device.
The organisation must have a documented procedure for issuing advisory notices in the event of such an occurrence, and it should be ready to be put into effect at any time. Therefore, the organisation cannot shut down completely for vacation for a week without having mechanisms in place to get hold of the necessary staff to run the process.
ISO 13485 requirements on CAPA – Corrective actions and preventive actions
CAPA stands for Corrective Action and Preventive Action and is essential to the ISO 13485 quality management system. The purpose of CAPA is to prevent nonconformities from recurring or occurring. CAPA is applicable to both nonconformities relating to medical devices but also to services and the quality management system.
Implementing corrective actions
Corrective actions are defined as actions to eliminate the cause of a nonconformity and to prevent recurrence of one. Do not confuse corrective actions with corrections; the difference being that a correction is a one-time solution applied to the problem at hand, while corrective actions prevent the same problem from happening again.
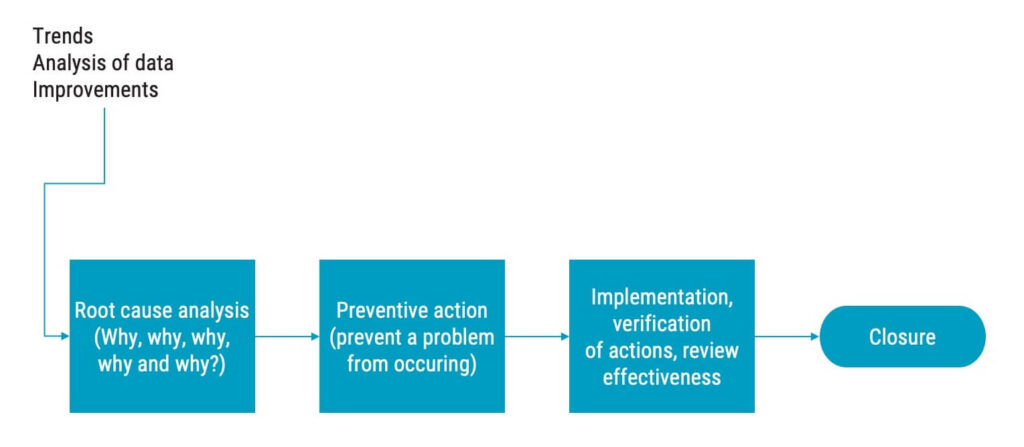
The first step of corrective actions is always to investigate why the nonconformity occurred. Based on the findings of this investigation, actions must be taken. This investigation is often referred to as root cause analysis, albeit the term is not mentioned in ISO 13485. ISO 13485 will only say “determining the causes of nonconformities”, but this should, in most cases, be understood as performing a root cause analysis.
If a detected nonconformity involves a low risk, then it might suffice to fix the issue through a small correction. However, if the risk is higher, ISO 13485 requires you to implement corrective actions that are proportionate to the effects of the nonconformities encountered.
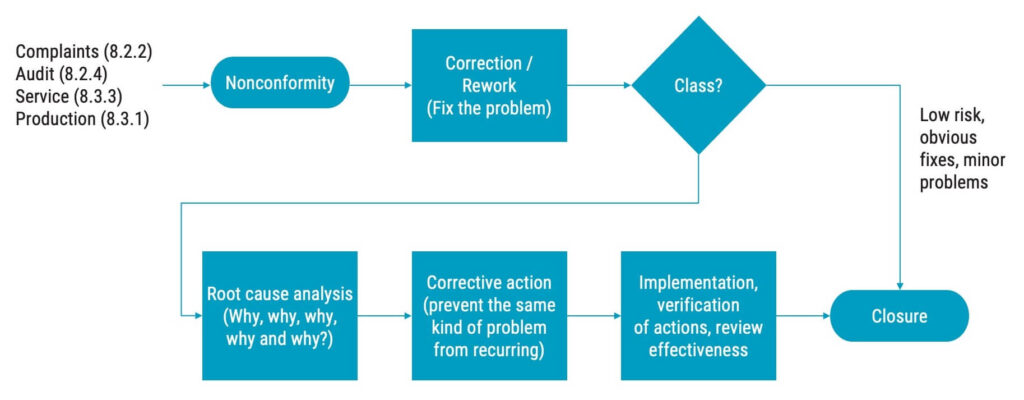
Your CAPA process must include how to determine the causes of nonconformities, which is most often done through a root cause analysis. One way to do that is to ask the question “Why?” five times. You could also do a qualitative analysis or use a fishbone diagram to determine the root cause.
Once you have determined the root cause, the corrective actions should be planned and documented before the implementation begins.
After implementation, you need to verify that the actions you take will not:
- Adversely affect the ability to meet regulatory requirements. or
- Adversely affect the safety and performance of the medical device.
You must also verify the effectiveness of the corrective actions to ensure that they achieve the results that you planned they would.
Implementing preventive actions
Preventive actions are similar to corrective actions, the difference is that preventive actions are initiated without a preceding nonconformity.
Preventive actions are about proactively preventing occurrence of potential nonconformities before something goes wrong. Preventive actions are typically initiated as the result of decisions made in management review.
ISO 13485 requirements on managing complaints
According to ISO 13485:2016, 3.4, complaints are:
Written, electronic or oral communication that alleges deficiencies related to the identity, quality, durability, reliability, usability, safety, or performance of a medical device that has been released from the organisation’s control or related to a service that affects the performance of such medical devices.
Anyone who comes in contact with any external people that are not part of the medical device organisation must know when they receive a complaint. This includes, for example, anyone who could potentially answer the phone at the organisation or meet with customers.
It should be noted that the QSR, uses a slightly different definition of complaint.

ISO 13485 requires medical device organisations to establish a documented procedure for receiving, evaluating, and investigating complaints. Furthermore, a complaint may have to be reported to regulatory authorities if it involves harm to people or potential harm to people. Records of complaints must be maintained. See section 8.2.2 in ISO 13485 for more details relating to complaints handling.
Summary: Working with quality management for medical devices and ISO 13485
Quality management and implementing an effective, as well as efficient QMS are crucial success factors for organisations working with medical devices. It involves systematic processes designed to ensure that a product consistently meets predefined requirements.
Understanding quality management for medical devices and ISO 13485 is much needed for anyone working in the medical device industry.
Would you like to know more about quality management?
If you want to know more about ISO 13485 and quality management for medical devices, take a look at our online Quality Management for Medical Devices and ISO13485 course. This comprehensive course is specifically tailored to make the requirements of the ISO 13485 as tangible and concrete as possible, so participants can confidently work in an organisation where ISO 13485 requirements apply. It introduces tools and methods on how to work successfully and efficiently within a quality management systems.
Or if you’re looking for a tailored training to align with your company’s specific needs – contact us for in-house training options.
Our online courses are frequently taken by competent authorities, notified bodies and medical device manufacturers and distributors.



