
Article 22
Systems and procedure packs
1. Natural or legal persons shall draw up a statement if they combine devices bearing a CE marking with the following other devices or products, in a manner that is compatible with the intended purpose of the devices or other products and within the limits of use specified by their manufacturers, in order to place them on the market as a system or procedure pack:
(a) other devices bearing the CE marking;
(b) in vitro diagnostic medical devices bearing the CE marking in conformity with Regulation (EU) 2017/746;
(c) other products which are in conformity with legislation that applies to those products only where they are used within a medical procedure or their presence in the system or procedure pack is otherwise justified.
2. In the statement made pursuant to paragraph 1, the natural or legal person concerned shall declare that:
(a) they verified the mutual compatibility of the devices and, if applicable other products, in accordance with the manufacturers’ instructions and have carried out their activities in accordance with those instructions;
(b) they packaged the system or procedure pack and supplied relevant information to users incorporating the information to be supplied by the manufacturers of the devices or other products which have been put together;
(c) the activity of combining devices and, if applicable, other products as a system or procedure pack was subject to appropriate methods of internal monitoring, verification and validation.
3. Any natural or legal person who sterilises systems or procedure packs referred to in paragraph 1 for the purpose of placing them on the market shall, at their choice, apply one of the procedures set out in Annex IX or the procedure set out in Part A of Annex XI. The application of those procedures and the involvement of the notified body shall be limited to the aspects of the procedure relating to ensuring sterility until the sterile packaging is opened or damaged.
The natural or legal person shall draw up a statement declaring that sterilisation has been carried out in accordance with the manufacturer’s instructions.
4. Where the system or procedure pack incorporates devices which do not bear the CE marking or where the chosen combination of devices is not compatible in view of their original intended purpose, or where the sterilisation has not been carried out in accordance with the manufacturer’s instructions, the system or procedure pack shall be treated as a device in its own right and shall be subject to the relevant conformity assessment procedure pursuant to Article 52.
The natural or legal person shall assume the obligations incumbent on manufacturers.
5. The systems or procedure packs referred to in paragraph 1 of this Article shall not themselves bear an additional CE marking but they shall bear the name, registered trade name or registered trade mark of the person referred to in paragraphs 1 and 3 of this Article as well as the address at which that person can be contacted, so that the person’s location can be established. Systems or procedure packs shall be accompanied by the information referred to in Section 23 of Annex I. The statement referred to in paragraph 2 of this Article shall be kept at the disposal of the competent authorities, after the system or procedure pack has been put together, for the period that is applicable under Article 10(8) to the devices that have been combined. Where those periods differ, the longest period shall apply.
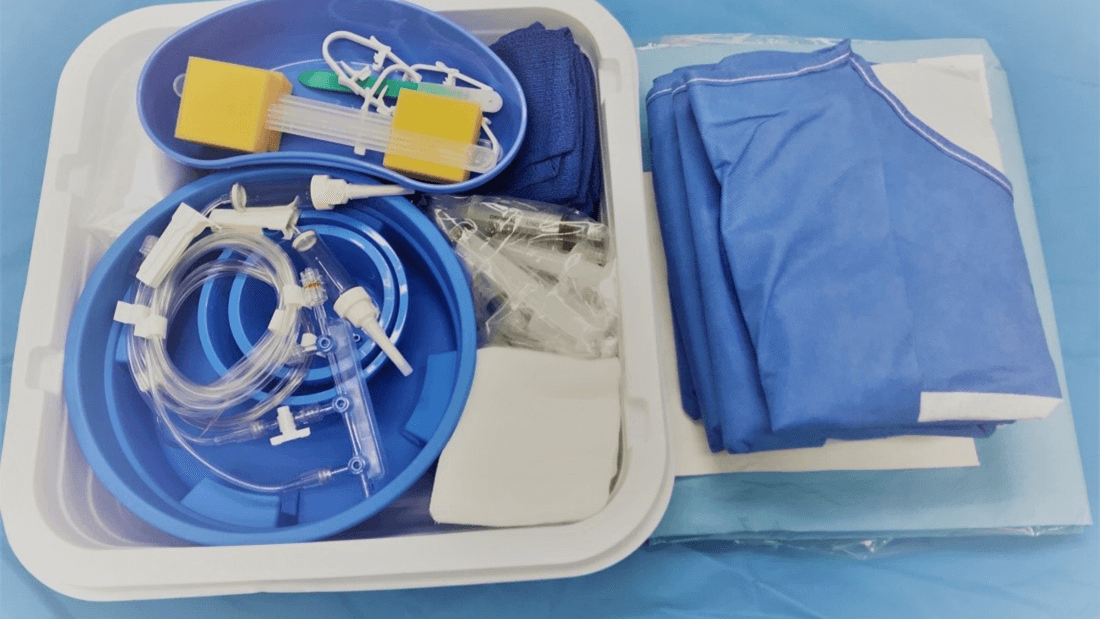
The picture shows what can typically be referred to as both system AND procedure pack. It meets the definitions of both. Let’s look at the definition of procedure packs and systems found in article 2 of the MDR.
(10) ‘procedure pack’ means a combination of products packaged together and placed on the market with the purpose of being used for a specific medical purpose;
(11) ‘system’ means a combination of products, either packaged together or not, which are intended to be inter- connected or combined to achieve a specific medical purpose;
In this image, we have a procedure pack made up of swabs, cups, and drapes to be used for a specific medical purpose or procedure. And it is also a system since syringes, distributors and tubing are needed in combination to achieve a specific medical purpose. But whether it is a procedure pack or a system is not that critical since the requirements are the same for both system and procedure packs. And both need to be registered in EUDAMED as devices.
So these products should only consist of already CE-marked products put together by an economic operator and placed on the market. No additional CE-mark shall be applied to those packs, but the name and address of the responsible economic operator must be stated on the pack. The Systems and procedure packs’ economic operator must draw up a statement that shall be made available to the competent authorities according to the requirements in section 2 of article 22 (found above). And if the economic operator cannot issue the statement, the system or procedure pack operator becomes the legal manufacturer and needs to treat the pack as a medical device in its own right.
To summarise this, you can say that the pack manufacturer needs to ensure that the compatibility of all devices has been verified and that sufficient information for use is supplied. Also, the pack manufacturer shall be able to show how it has done this in a controlled way with evidence of verification and validation.
If the systems or procedure pack is sterilised after put together, the manufacturer of the sterile systems or procedure pack shall basically follow the same rules as for any class I sterile devices, independent from the class of the devices included in the pack. And this assumes that the included devices are intended for the chosen sterilisation process according to the original legal manufacturers. Otherwise, it becomes a medical device in its own right and shall be treated as such.

Pontus Gedda is a dedicated medical device specialist that has worked both in the industry, as a design engineer and project manager, and in the notified body world as a medical device lead auditor and manager.
He has vast experience in the MDR and its implementation thru hands-on experience from implementing the MDR at a notified body and leading that notified body through a joint assessment and getting designated as an MDR notified body.
He was a senior manager at a notified body during the transition from MDD to MDR and also a member of the NB-MED group.

Receive FREE templates and quarterly updates on upcoming courses that can help you in your career! Subscribe to our newsletter now.
When you submit this form, you will be sending personal information to medicaldevicehq.com. To comply with GDPR requirements, we need your consent to store and use the personal data you submit. Take a look at our Privacy policy for more details.
Once you have submitted the form, you will be automatically taken to your cart where the e-book and 100% discount will be applied. Go through checkout to get the free e-book.
Press here to subscribe to our newsletter and get your free e-book