
This article aims to answer the question of what is a medical device, and who is responsible for it, according to the MDR.
The following video is a topic from our online course Introduction to MDR, Medical Device Regulation (EU) 2017/745, and you can register for it by clicking the link.
It is important to know if a product is a medical device because this is what determines if the MDR applies to that product or not. Therefore, it has a major impact on how the device realisation is done as well as how the product should be approved for the European market.
The simplest way of describing what a medical device is would be to say that medical devices are things found in hospitals for treatment that are not medicines. Still, it is not that simple. Medical devices can be found everywhere, not only in hospitals; they can be bought in a pharmacy, but also in a supermarket, like band-aids.
In the MDR, a medical device is stated in Article 2, “Definitions”, as the very first definition and it includes specific medical purposes such as diagnosis, monitoring, treatment, alleviation, and other purposes.
One important aspect that determines if a product is a medical device or not is whether it is intended to treat or monitor a patient, whilst not being a ‘drug’. The treatment can happen in different ways, and the complete definition can be found in Article 2 of the Medical Device Regulation.
It is not only tangible products that can be touched that could be medical devices; stand-alone software or mobile apps can be too.
In order to determine whether a product is a medical device or not, the intended purpose, or intended use needs to be known. This intended purpose is what the legal manufacturer of the product claims that the product is intended to be used for.
So, if the manufacturer is planning on selling something to the EU with an intended purpose that falls within the definition of a medical device, the product should also be regulated and handled as a medical device.
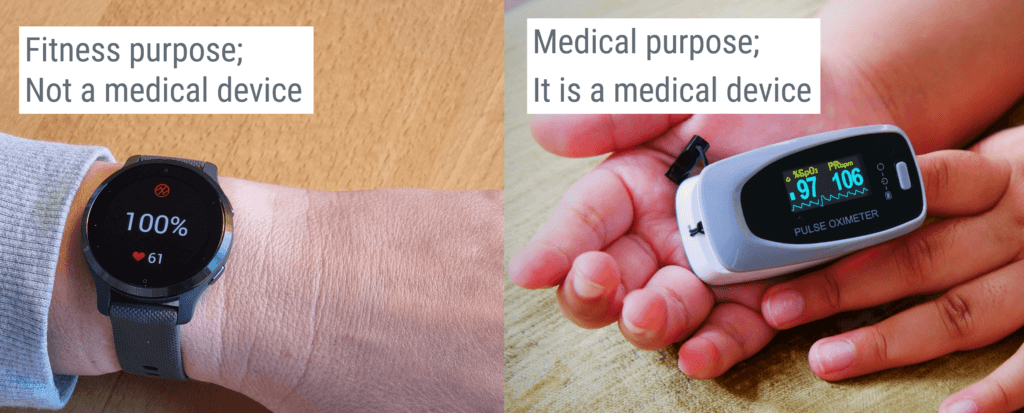
For example, if the product is a watch with a built-in oxygen saturation measurement, but it is intended to be used for fitness purposes only, it will not be classified as a medical device since it is not to be used for diagnosis or treatment. However, if it is intended to be used for monitoring a person’s oxygen saturation for medical or therapeutic purposes, it becomes a medical device.
The MDR defines intended purpose as
“the use for which a device is intended according to the data supplied by the manufacturer on the label, in the instructions for use or in promotional or sales materials or statements and as specified by the manufacturer in the clinical evaluation.”
MDR, Article 2, 12
Clinical evaluation is a central concept in the MDR. It is the process by which the manufacturer evaluates relevant clinical data to ensure that the medical device performs within its intended purpose and that it is safe and effective. The outcome of the clinical evaluation is documented in a clinical evaluation report. It is essential to differentiate between clinical evaluation and clinical investigation, where the latter is performed on human subjects.
The intended purpose should not only be a statement in the instructions for use; it also needs to be completely aligned with what the manufacturer says on the labels, and in the promotional or sales materials, or other statements made by the company.
Therefore, it is of utmost importance that the manufacturer is stringent in the definition of the intended purpose of the device, both before and after putting it on the market.
Oftentimes, it is fairly easy to decide whether a product should be a medical device or not. But sometimes, it is not that obvious. When it is not obvious, it is highly recommended to read the exact MDR definition word by word and also look into the classification annex found at the end of MDR, which is in this case the Annex 8. This annex provides an insight into how medical devices can be used.
There are also guidance documents, for example the Borderline manual that was introduced under the MDD that can be used. It is not fully applicable to the MDR, but the principles from the Borderline document could be used and extrapolated to what it would be within the MDR.
Some products are obvious medical devices by themselves, but it can be difficult to define products that are used in conjunction with a medical device. They are referred to as accessories, and they are also regulated as medical devices, so the same requirements apply.
Sometimes it is hard to determine what is a medical device and what is an accessory, and there are different views on this, but since the same requirements apply to both, it is not that crucial. In the worst case, the manufacturer and the Notified body discuss what is the medical device and what is the accessory.
Taking an IV drip chamber as an example, there is a roller clamp used together with the IV drip chamber on the drip line. So, both of these are medical devices that can be used in combination or potentially separately. But before the IV drip chamber works, it needs to be elevated by hanging it on a stand so the IV fluid actually reaches the patient.
That stand is needed to fulfil the intended use of the drip chamber and is, therefore, an accessory to that medical device. Accessories that are needed to fulfil a medical device’s intended purpose are also medical devices.
But the tricky part is the question – is the roller clamp an accessory? The answer is that it is probably not, since it is normally used for an IV drip and potentially other medical device applications, so it is a combination. The stand, on the other hand, will more likely not have several other medical uses, so it should probably be seen as an accessory.

Accessories shall be classified in their own rights, independent of what they are used together with. This applies to a combination of products as well, which are classified in their own rights too.
Some medical devices are delivered as systems or procedure packs. You can read more about this in article 22 of the MDR.
Some other products shall also be deemed to be medical devices, such as devices for the control or support of conception. Products specifically intended for the cleaning, disinfection, or sterilisation of medical devices are in themselves also regulated as medical devices.
There is a special group of products that are not medical devices by definition, which means that they do not fall within the MDR’s definition of what a medical device is, but the MDR still applies to them. These are products that could be implanted, invasive, or harmful, often used only for cosmetic reasons or at least without a medical purpose. These were not previously regulated under the MDD, but they now must meet the same requirements as if they were medical devices.
Examples of such products, without a medical purpose, from Annex XVI are: coloured contact lenses, surgically invasive products without medical purpose (excluding piercings and tattoos), dermal fillers, liposuction equipment, and equipment for removal of hair and tattoos using light, infrared light or laser.
And lastly, an interesting thing to mention is that prevention for injury or disability is not included in the definition of a medical device since that would make the device a Personal Protection Equipment (PPE) instead, and there is another directive for that kind of equipment.
Please note that this was all for the definition of a medical device in the European Union. There could be other definitions of medical devices on other markets around the world, meaning that a product that is considered a medical device in the US might not be considered a medical device in the EU. It is not the country of the manufacturer that states what is a medical device, but the legislation of the country where the product is sold.
It is worth mentioning that the MDR has replaced both of the former directives for medical devices, the MDD and AIMDD. AIMDD was the directive for Active Implantable Medical Devices so those are now also included in the MDR, meaning that there is only one regulation for all medical devices in Europe.

Another new thing in the MDR is that for most implants the manufacturer also needs to supply all products with an implant card (according to article 18). The hospital implanting the device then needs to fill in and give the implant card to the patient receiving the implant.
The manufacturer needs to supply a SSCP to the patient for all implants. You can read more about this under article 32.
It is very clear that the manufacturer (or sometimes the legal manufacturer) is the one responsible for the medical device according to the MDR. This is the natural or legal person that is responsible for the medical device both before and after putting it on the market, by indicating their name or trademark on the device. The manufacturer does not necessarily produce the product, which is a crucial concept since many medical devices are produced by subcontractors. But it is nevertheless the manufacturer that is responsible for the device, and this is the operator to be identified on the medical device labelling. This is for the regulatory authorities to be able to say which organisation is ultimately responsible for a medical device on the market.
Who the legal manufacturer is must always be very clear on the labels. This should primarily be stated on the device itself, but if not possible, at least on the label, packaging material, and accompanying documentation. If the device is just labeled with trademarks, the actual name of the legal manufacturer still needs to be indicated.
In conclusion, learning and ultimately deciding what constitutes a medical device is not always easy, but carefully following the MDR leaves no space for mistakes.
If you enjoyed this article, make sure to take a look at the ones we previously published here.
If you want to know more about understanding the European regulation for medical devices, take a look at our online Medical Device Regulation (EU) 2017/745 course. This online course is an in-depth overview of the Medical Device Regulation according to (EU) 2017/745 as well as related guidances, like MDCG, and how to apply to a notified body for conformity assessment.
It is suitable for anyone working with regulatory questions, such as RA and QA engineers, PRRC or management.

Pontus Gedda is a dedicated medical device specialist that has worked both in the industry, as a design engineer and project manager, and in the notified body world as a medical device lead auditor and manager.
He has vast experience in the MDR and its implementation thru hands-on experience from implementing the MDR at a notified body and leading that notified body through a joint assessment and getting designated as an MDR notified body.
He was a senior manager at a notified body during the transition from MDD to MDR and also a member of the NB-MED group.

Receive FREE templates and quarterly updates on upcoming courses that can help you in your career! Subscribe to our newsletter now.
When you submit this form, you will be sending personal information to medicaldevicehq.com. To comply with GDPR requirements, we need your consent to store and use the personal data you submit. Take a look at our Privacy policy for more details.

