
As the final part of this series of three, this article will focus on categorising risks according to the different life-cycle phases of a medical device to efficiently address them to avoid harm. Read Part 1 about risk assessment here and Part 2 here.
While not all manufacturers may segment their documentation into three distinct documents as per Team NB’s recommendation, risk association with life-cycle phases is a common practice in the industry. Therefore, this information is likely to hold relevance for most manufacturers.
Let’s reuse the established sequence of life-cycle phases from the second article, to illustrate how various risks can be associated with life-cycle phases.
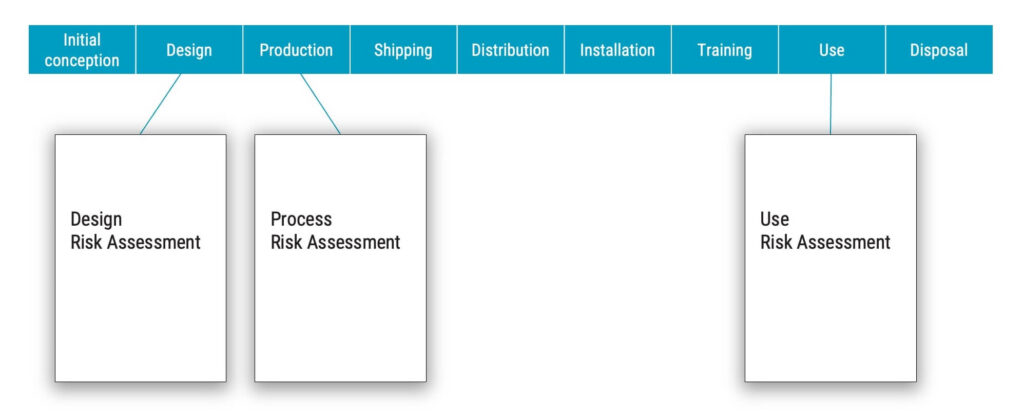
In the realm of risk management for medical devices, the majority of risks become harms during the use phase. However, it is also crucial to recognise that harms are not confined to the use phase alone. Indeed, harm could emerge during other stages of a device’s life cycle, such as installation, training, or even disposal.
The heat map below illustrates when most harms would occur with the majority of medical devices. Harms can, of course, also occur during production to production personnel, but this is usually not part of medical device risk management but should be part of your workplace safety procedures.

Assuming that most harms materialise in the life-cycle phases marked in yellow and orange, the question is: In which life-cycle phase can you find the root cause of the risk that will later result in harm?
The root cause can be traced back to initial conception and design. It is in these phases where manufacturers “create risk” by, for example, choosing a poor design, not performing proper verification, or making bad decisions regarding the production process.
One could argue that many risks are created in the use phase as well when there are use errors. While that is true to some degree, it has not been included because use errors that lead to hazardous situations with unacceptable risk should already have been mitigated by the design as part of the usability engineering and risk management processes.
So, when there is a use error that results in a hazardous situation, it is still a failure in the design phase, and therefore, the root cause is in the initial conception or design phase.
Decisions regarding risk control measures are made during the initial conception and design phases. This is where manufacturers for example:
These decisions are tightly connected with when the risk control measures are implemented, which is in the design and production phases. From this point onwards, they remain effective.
For example, you may have decided on certain shipping requirements to avoid damaging the medical device, and determined that mandatory in-service training is required before a customer can use the medical device.

The reasonably foreseeable sequences or combinations of events take place throughout all life-cycle phases, which leads us to the most important question: What criteria do you use to determine what life-cycle phase a risk belongs to?
If the criteria state that when harm occurs, essentially all your risks would be found in the use risk assessment document. However, if you are truly going to the root cause of these risks, then many of the risks will be found in the design risk assessment document.
It’s crucial for medical device manufacturers to conduct a thorough review of their procedures and risk management plans to ensure that there are clear criteria in place to identify which life-cycle phases a risk belongs to.
By doing this, potential risks can be identified and mitigated at the earliest possible stage, thereby ensuring the integrity and safety of the medical devices being produced.
Ask yourself what criteria determine which life-cycle phase or document a risk belongs to?
The recommended approach is to associate the risks with life-cycle phases where sequences or combinations of events take place. This means that most risks can be associated with more than one life-cycle phase. And this in turn means that if the risk management documents are divided based on life-cycle phases, it will be challenging to determine which document the risk should be recorded in, or even worse, the decision to allocate the risk to a particular document is arbitrary.
It is important to remember that a sequence of events can come from many phases. It can start with a poor design, which is then further amplified by production and shipping, and then events take place in the use phase that eventually result in a hazardous situation and harm.

It is your job as the manufacturer to identify the association with all the relevant life-cycle phases to accurately determine the true root cause of the risk to reduce and control it as effectively as possible.
Ensure that your organisation has criteria for how you define and determine which life-cycle phases you associate risks with as part of your risk management system.
And a final reminder, always go for the root cause of the risk – You are required to do so, and you will sleep a lot better if you do.
Get instant access to our online Risk Management for Medical Devices and ISO 14971:2019 course right here. In 10 hours, you can learn more about how to develop new medical devices and maintain them in organisations where design control requirements apply. This course is taken by quality assurance, project management, design engineering or those involved in R&D and product development teams.

Peter Sebelius is a highly esteemed trainer, consultant and entrepreneur in the medical device industry. He is a member of the Joint Working Group that is revising the ISO 13485 and ISO 14971 standards.
He has vast ‘hands on’ experience, having developed, amongst other things, a mechanical chest compression device and an ex vivo perfusion machine for lungs. He has received numerous awards including the Great Design Award and the title “This year’s specialist” by Veckans affärer.

Receive FREE templates and quarterly updates on upcoming courses that can help you in your career! Subscribe to our newsletter now.
When you submit this form, you will be sending personal information to medicaldevicehq.com. To comply with GDPR requirements, we need your consent to store and use the personal data you submit. Take a look at our Privacy policy for more details.
Special launch offer: 349 299 EUR for the online plan & 449 349 EUR for the online lifetime plan.

