
When placing a medical device or in-vitro diagnostic medical device on the EU market, it is mandatory to fulfil the General Safety and Performance Requirements (GSPR) from Annex I of the Medical Device Regulation and In-Vitro Diagnostic Medical Device Regulation. Keep reading to learn more about them, and make sure you check out our other articles on similar topics from the medical device industry.
Also, this article deals with a topic you can learn more about in our online Design control for medical devices course, but feel free to browse through our other courses, too.
The GSPR should be considered in the planning phase. More importantly, they should be brought up very early in the planning phase because you cannot plan a budget or schedule if you are unaware of the requirements that apply to your medical device.

The GSPR take up 14 pages of the MDR. The General Safety and Performance Requirements include the following main categories of requirements:
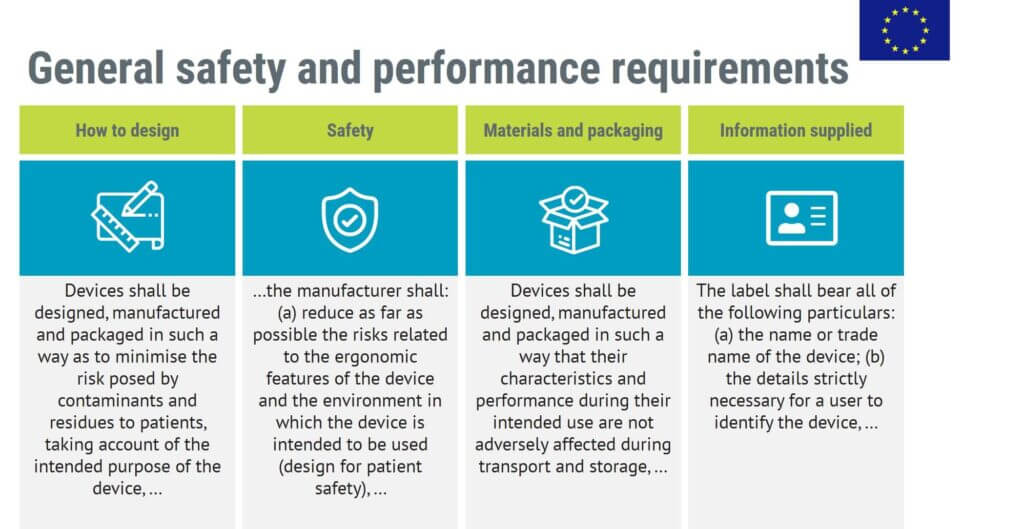
General Safety and Performance Requirements are obligatory. You have to comply with the GSPR that apply to your medical device if you are to place your medical device on the EU market.
The GSPR are sometimes written on a granular level. This is, for example, true regarding the requirements for information supplied by the manufacturer.
But still, many GSPR are high-level requirements, which means it can be challenging to understand how to implement them.
To ensure you fulfil the GSPR, you are recommended to use harmonised standards.
Harmonised standards are standards that have been designated as harmonised by the EU. It means that if your medical device and quality management system meet the requirements of the harmonised standards, you can presume conformity with the General Safety and Performance Requirements that the standard covers.
For example, when the GSPR requires you to implement a risk management system, you can turn to the harmonised standard ISO 14971 on risk management to find more granular and up-to-date requirements for a risk management system.
Harmonised standards offer two important advantages:
1. They are more granular than the GSPR, thereby making it easier to understand how to fulfil the requirements
2. They are updated more frequently and, therefore, more up to date. Most standards have a 5-year review cycle, whereas the Medical Device Regulation might be around for 15–30 years. With rapid technological development, the standards can better keep up the pace than the regulations can.
As the requirements of the harmonised standards are building up to meet the GSPR, you really should meet the requirements of any applicable harmonised standards to avoid problems with the regulatory approvals.
The harmonised standards are not mandatory per se, as they are standards, not regulations. But they are as close to mandatory as they can get. Every regulatory body will expect you to conform to applicable harmonised standards. Not doing so can cost a lot of time and money while arguing why your quality management system or medical device are meeting the requirements of the MDR while not fulfilling the requirements of harmonised standards.
And even though you may eventually achieve regulatory approval without fulfilling the requirements of the harmonised standards, it is a risky path.
At the time of the authoring of this article, the following standards are harmonised with the Medical Device Regulation:
| Harmonised standards | |
|---|---|
|
EN 285:2015+A1:2021
|
Sterilization - Steam sterilizers - Large sterilizers
|
|
EN ISO 10993-9:2021
|
Biological evaluation of medical devices - Part 9: Framework for identification and quantification of potential degradation products (ISO 10993-9:2019)
|
|
EN ISO 10993-10:2023
|
Biological evaluation of medical devices - Part 10: Tests for skin sensitization (ISO 10993-10:2021)
|
|
EN ISO 10993-12:2021
|
Biological evaluation of medical devices - Part 12: Sample preparation and reference materials (ISO 10993-12:2021)
|
|
EN ISO 10993-23:2021
|
Biological evaluation of medical devices - Part 23: Tests for irritation (ISO 10993-23:2021)
|
|
EN ISO 11135:2014,
EN ISO 11135:2014/A1:2019
|
Sterilization of health-care products - Ethylene oxide - Requirements for the development, validation and routine control of a sterilization process for medical devices (ISO 11135:2014)
|
|
EN ISO 11137-1:2015,
EN ISO 11137-1:2015/A2:2019
|
Sterilization of health care products - Radiation - Part 1: Requirements for development, validation and routine control of a sterilization process for medical devices (ISO 11137-1:2006, including Amd 1:2013)
|
|
EN ISO 11737-1:2018,
EN ISO 11737-1:2018/A1:2021
|
Sterilization of health care products - Microbiological methods - Part 1: Determination of a population of microorganisms on products (ISO 11737-1:2018)
|
|
EN ISO 11737-2:2020
|
Sterilization of health care products - Microbiological methods - Part 2: Tests of sterility performed in the definition, validation and maintenance of a sterilization process (ISO 11737-2:2019)
|
|
EN ISO 13408-6:2021
|
Aseptic processing of health care products - Part 6: Isolator systems (ISO 13408-6:2021)
|
|
EN ISO 13485:2016,
EN ISO 13485:2016/AC:2018,
EN ISO 13485:2016/A11:2021
|
Medical devices - Quality management systems - Requirements for regulatory purposes (ISO 13485:2016)
|
|
EN ISO 14160:2021
|
Sterilization of health care products - Liquid chemical sterilizing agents for single-use medical devices utilizing animal tissues and their derivatives - Requirements for characterization, development, validation and routine control of a sterilization process for medical devices (ISO 14160:2020)
|
|
EN ISO 14971:2019,
EN ISO 14971:2019/A11:2021
|
Medical devices - Application of risk management to medical devices (ISO 14971:2019)
|
|
EN ISO 15223-1:2021
|
Medical devices - Symbols to be used with information to be supplied by the manufacturer - Part 1: General requirements (ISO 15223-1:2021)
|
|
EN ISO 17664-1:2021
|
Processing of health care products - Information to be provided by the medical device manufacturer for the processing of medical devices - Part 1: Critical and semi-critical medical devices (ISO 17664-1:2021)
|
|
EN ISO 25424:2019
|
Sterilization of health care products - Low temperature steam and formaldehyde - Requirements for development, validation and routine control of a sterilization process for medical devices (ISO 25424:2018)
|
|
EN ISO 25424:2019,
EN ISO 25424:2019/A1:2022
|
Sterilization of health care products - Low temperature steam and formaldehyde - Requirements for development, validation and routine control of a sterilization process for medical devices (ISO 25424:2018)
|
|
EN IEC 60601-2-83:2020,
EN IEC 60601-2-83:2020/A11:2021
|
Medical electrical equipment - Part 2-83: Particular requirements for the basic safety and essential performance of home light therapy equipment
|
To ensure you are using the latest list of harmonised standards, use the following links to check the current status:
Medical Device Regulation (click the document at the end of the page)
In-Vitro Diagnostic Medical Device Regulation (click the document at the end of the page)
Common specifications are closely related to General Safety and Performance Requirements. A common specification is a standard that has been created based on an initiative from the EU, and not through the regular standardisation process from ISO and IEC.
The MDR defines common specifications as:
A set of technical and/or clinical requirements, other than a standard, that provides a means of complying with the legal obligations applicable to a device, process or system.
When there are no appropriate harmonised standards, or they are insufficient, and when there is a need to address public health concerns, the European Commission may adopt common specifications.
They can be adopted with respect to technical documentation from Annexes II and III, clinical evaluation and post-market clinical follow-up from Annex XIV, or the requirements regarding clinical investigation from Annex XV.
The common specifications are not mandatory per se, but the same principle applies as with harmonised standards: you should meet their requirements.
The only situation where you can skip them is if you can justify the reason while simultaneously ensuring the equivalent level of safety and performance.

To conclude, GSPR are obligatory, harmonised standards and common specifications are as close to mandatory as they can get without actually being mandatory.
The MDR and IVDR require that the technical documentation contains information that shows that the General Safety and Performance Requirements have been met. This is usually done by filling out a GSPR checklist. The checklist should:
1. Show which GSPR apply to the medical device,
2. Provide an explanation to why others don’t apply,
3. Methods used to demonstrate conformity with the applicable GSPR,
4. Harmonised standards and common specifications that have been applied, and
5. Reference to the documents that shows evidence of conformity with each harmonised standard or common specification.
Below is an example of a template for a GSPR checklist:

General Safety and Performance Requirements is a EU concept, but what about the US market? Are there similar requirements? Yes, there are, however, they are not as pronounced as in the EU where the GSPR are part of the regulations.
The corresponding requirements for the US market would be the Essential Principles of Safety and Performance of Medical Devices and IVD Medical Devices that are published by the International Medical Device Regulators Forum. But again, the essential principles are not implemented as a regulation, thus they would have a weaker standing in the US compared to the GSPR in the EU.
If you want to know more about understanding the European regulation for medical devices, take a look at our online Medical Device Regulation (EU) 2017/745 course. This online course is an in-depth overview of the Medical Device Regulation according to (EU) 2017/745 as well as related guidances, like MDCG, and how to apply to a notified body for conformity assessment.
It is suitable for anyone working with regulatory questions, such as RA and QA engineers, PRRC or management.

Pontus Gedda is a dedicated medical device specialist that has worked both in the industry, as a design engineer and project manager, and in the notified body world as a medical device lead auditor and manager.
He has vast experience in the MDR and its implementation thru hands-on experience from implementing the MDR at a notified body and leading that notified body through a joint assessment and getting designated as an MDR notified body.
He was a senior manager at a notified body during the transition from MDD to MDR and also a member of the NB-MED group.

Receive FREE templates and quarterly updates on upcoming courses that can help you in your career! Subscribe to our newsletter now.
When you submit this form, you will be sending personal information to medicaldevicehq.com. To comply with GDPR requirements, we need your consent to store and use the personal data you submit. Take a look at our Privacy policy for more details.
Once you have submitted the form, you will be automatically taken to your cart where the e-book and 100% discount will be applied. Go through checkout to get the free e-book.
Press here to subscribe to our newsletter and get your free e-book
Special launch offer: 349 299 EUR for the online plan & 449 349 EUR for the online lifetime plan.

