
Das Qualitätsmanagement für Medizinprodukte bezieht sich auf die Systeme und Prozesse, mit denen sichergestellt wird, dass Medizinprodukte sicher und wirksam sind und die gesetzlichen Anforderungen erfüllen.
Die Norm ISO 13485 enthält Anforderungen für das Management des gesamten Lebenszyklus eines Medizinprodukts, von der Konzeption und Entwicklung über die Herstellung und den Vertrieb bis hin zur Überwachung nach dem Inverkehrbringen.
Dieser illustrierte Leitfaden wurde von Peter Sebelius verfasst, einem Mitglied der Arbeitsgruppe, die die neuesten Versionen von ISO 13485 und ISO 14971 verfasst hat.
Dieser Leitfaden wird:
Qualitätsmanagement ist nicht nur wichtig, um Produkte zu verkaufen, die die Kundenanforderungen erfüllen oder übertreffen. In der Medizinprodukteindustrie ist das Qualitätsmanagement auch wichtig für die Einhaltung von Vorschriften und den Marktzugang.
Medizinproduktehersteller müssen verschiedene Vorschriften einhalten, um sicherzustellen, dass ihre Produkte bestimmte Qualitätsanforderungen erfüllen. Besonders hervorzuheben ist die Norm ISO 13485 für Qualitätsmanagement. Die ISO 13485 ist in diesem Bereich wichtig, wenn nicht sogar die wichtigste Norm, und kann zur Erfüllung der Anforderungen der Vorschriften verwendet werden.
Wenn Definitionen von Schlüsselbegriffen in der ISO 13485 fehlen, sollte man nicht primär zu Wörterbüchern greifen, sondern die ISO 9000:2015 Qualitätsmanagementsysteme – Grundlagen und Begriffe lesen.
In ISO 9000:2015 wird Qualitätsmanagement als “Management in Bezug auf Qualität” definiert. Diese Definition ist nicht besonders nützlich, aber wenn die Begriffe aus der Definition erweitert werden, ergibt sie mehr Sinn.

Qualitätsmanagement kann als Koordinierung der Aktivitäten zur Steuerung und Kontrolle einer Organisation im Hinblick auf die Erfüllung der Anforderungen an ein Produkt, eine Dienstleistung, einen Prozess oder ein System gesehen werden.
Erfolgreiches Qualitätsmanagement ist ohne ein Qualitätsmanagementsystem (QMS) praktisch unmöglich.
Ein QMS ist eine Reihe von Strategien, Prozessen und Verfahren, die von einer Organisation mit dem Hauptziel des Qualitätsmanagements umgesetzt werden.
Qualitätsmanagementsysteme können entweder papiergestützt oder elektronisch sein. Jede Art von System bringt ihre eigenen Vorteile und Herausforderungen mit sich.
Bei einem papiergestützten System handelt es sich in der Regel um eine Reihe von Dokumenten, die sich in Ordnern befinden, oder um gescannte Versionen von signierten Papierkopien, auf die über ein internes Netzwerk zugegriffen werden kann.
Ein papiergestütztes QMS ist einfach zu implementieren und erfordert keine großen Anfangsinvestitionen, aber wenn eine Organisation wächst, sind papiergestützte Systeme anfälliger für menschliche Fehler und langsamer.
Ein elektronisches QMS oder eQMS enthält in der Regel Verfahren und Vorlagen als elektronische Dokumente, die vollständig elektronisch genehmigt und verteilt werden.
Ein eQMS ist oft schneller und effizienter und kann das Fehlerrisiko verringern, aber die Einführung kann eine erhebliche Investition erfordern.
Ein hybrides QMS nutzt das Beste aus den beiden Welten des papiergestützten QMS und des eQMS und verwendet eine Mischung aus papiergestützten und elektronischen Dokumenten.
Alle Medizinprodukte, die in der Europäischen Union auf den Markt gebracht werden, müssen die gesetzlichen Anforderungen der EU erfüllen. Dies wird durch die Verordnung über Medizinprodukte (MDR) und die Verordnung über In-vitro-Diagnostika (IVDR) vorgeschrieben.
Abschnitt 9 in Artikel 10 der MDR und IVDR legen eindeutig fest, dass die Herstellerorganisationen ein Qualitätsmanagementsystem einrichten, dokumentieren, umsetzen, aufrechterhalten, auf dem neuesten Stand halten und kontinuierlich verbessern müssen.
Das QMS trägt wesentlich dazu bei, dass die Entwicklungs-, Herstellungs- und sonstigen Betriebsabläufe die Qualitäts- und Sicherheitsanforderungen erfüllen, so dass sichere, wirksame und den Vorschriften entsprechende Medizinprodukte entstehen.
Während die MDR und die IVDR die Anforderungen an das Qualitätsmanagement auf hohem Niveau festschreiben, geht die ISO 13485 viel tiefer ins Detail. Die ISO 13485-Norm ist 70 Seiten lang, im Gegensatz zur MDR, die insgesamt etwa 170 Seiten umfasst, aber nur etwa eine Seite mit Anforderungen speziell an das Qualitätsmanagementsystem enthält.
Die ISO 13485 ist mit der MDR und der IVDR harmonisiert. Die Harmonisierung der ISO 13485 bedeutet, dass, wenn die Anforderungen der ISO 13485 erfüllt sind, davon ausgegangen werden kann, dass die entsprechenden Anforderungen der MDR und IVDR ebenfalls erfüllt sind.

Die Einhaltung harmonisierter Normen, wie z. B. ISO 13485, ist zwar nicht ausdrücklich vorgeschrieben, aber durchaus wünschenswert. Es ist wichtig zu beachten, dass jede Abweichung von ISO 13485 oder anderen anwendbaren harmonisierten Normen eine solide Begründung erfordert.
Weitere Informationen darüber, wo Sie die ISO 13485-Norm herunterladen können, finden Sie hier.
In der EU gibt es zwei verschiedene Verordnungen für Medizinprodukte (MDR) bzw. In-vitro-Diagnostika (IVDR), in den Vereinigten Staaten unterliegen sowohl Medizinprodukte als auch In-vitro-Diagnostika der gleichen Verordnung, der 21 CFR 820 oder Quality System Regulation (QSR).
Die Food and Drug Administration (FDA) arbeitet aktiv an der Angleichung der Qualitätssystem-Verordnung an die ISO 13485. Die daraus resultierende Verordnung wird, sobald sie umgesetzt ist, als Qualitätsmanagementsystem-Verordnung (QMSR) bezeichnet. Die neuesten Informationen über die QMSR finden Sie hier.
Das Äquivalent zu harmonisierten Normen im US-Kontext sind anerkannte Konsensnormen. Die ISO 13485 ist keine anerkannte Konsensnorm für den US-Markt, da der QSR Anforderungen in denselben Bereichen wie die ISO 13485 enthält. Damit ist für den US amerikanischen Markt die QSR das entscheidende Dokument für die Erfüllung der Qualitätsanforderungen.
Das QMS muss in einer Weise dokumentiert und umgesetzt werden, die der Größe und Komplexität des Unternehmens und der Art der hergestellten Medizinprodukte angemessen ist. Die Norm ISO 13485 kann von einem Ein-Personen-Unternehmen bis hin zu einem multinationalen Unternehmen mit Tausenden von Mitarbeitern angewendet werden.
Die ISO 13485 kann auch auf Unternehmen mit sehr unterschiedlichen Tätigkeitsbereichen angewandt werden; so kann es sich zum Beispiel um ein Beratungsunternehmen oder einen Hersteller komplexer medizinischer Geräte handeln. Das bedeutet, dass es nicht nur eine akzeptable Lösung oder ein einheitliches QMS gibt, das die Anforderungen der ISO 13485 erfüllt. Im Gegenteil, es gibt unendlich viele Möglichkeiten, die Anforderungen der ISO 13485 umzusetzen.
Das QMS sollte keine statische Sammlung von Dokumenten sein. Es muss regelmäßig überprüft und aktualisiert werden, um seine kontinuierliche Wirksamkeit zu gewährleisten.
Organisationen sind verpflichtet, ein Qualitätshandbuch zu erstellen. Das Qualitätshandbuch ist das wichtigste Dokument für das QMS und der Ausgangspunkt für den Zugang zum Qualitätsmanagementsystem (QMS).
Das Qualitätshandbuch muss die Struktur der im Qualitätsmanagementsystem verwendeten Unterlagen darlegen. Zur Erfüllung dieser Anforderung wird in der Regel eine Übersicht verwendet, die der nachstehenden ähnlich ist, jedoch ohne die erläuternden Texte.

Darüber hinaus schreibt die ISO 13485 31 dokumentierte Verfahren vor. Diese dokumentierten Verfahren sind die Säulen Ihres Qualitätsmanagementsystems (QMS). Bei den 31 dokumentierten Verfahren muss es sich nicht unbedingt um 31 physische Dokumente handeln, sondern ein dokumentiertes Verfahren kann aufgeteilt werden und sich in mehreren Dokumenten wiederfinden. Oder mehrere dokumentierte Verfahren können in einem einzigen physischen Dokument zusammengeführt werden.
Die erforderlichen dokumentierten Verfahren sind in der Regel in Standardarbeitsanweisungen oder kurz SOPs zu finden. SOPs dienen als Handbücher, Anweisungen und Schulungsmaterial der Organisation, die den Mitarbeitern vorschreiben, wie sie ihre Arbeit auszuführen haben.
|
4.1.6 Validation of the application of computer software used in the quality management system
|
7.5.11 Preserving conformity of product to requirements during processing, storage, handling, and distribution
|
|
4.2.4 Controls needed to review, approve documents, update and re-approve documents
|
7.6 Procedures to ensure monitoring and measurement is done according to requirements
|
|
4.2.5 Controls needed for the identification, storage, security and integrity, retrieval, retention time and disposition of records
|
7.6 Validation of computer software used for monitoring and measurement
|
|
5.6.1 Management review
|
7.6 Calibration and verification
|
|
6.4.1 Monitor and control work environment if it has effect on product quality
|
8.2.1 Feedback
|
|
7.3.1 Design and development
|
8.2.2 Complaints handling
|
|
7.3.8 Transfer of design and development outputs to manufacturing
|
8.2.3 Notification of adverse events or issuance of advisory notices
|
|
7.3.9 Control of design and development changes
|
8.2.4 Responsibilities and requirements for planning and conduct of audits
|
|
7.4.1 Ensuring purchased product conforms to purchasing information
|
8.2.6 Monitoring and measuring characteristics of the product
|
|
7.5.1 Procedures for control of production
|
8.3.1 Responsibilities and authorities for identification, documentation, segregation, evaluation, and disposition of nonconforming product
|
|
7.5.6 Validation of processes
|
8.3.3 Issuing advisory notices
|
|
7.5.6 Validation of computer software used in production and service
|
8.3.4 Rework
|
|
7.5.7 Validation of processes for sterilization and sterile barrier systems
|
8.4 Collect and analyze appropriate data
|
|
7.5.8 Identification of production by suitable means through product realization
|
8.5.2 Reviewing nonconformities
|
|
7.5.8 Returned medical devices
|
8.5.3 Determining nonconformities and their causes
|
|
7.5.9.1 Traceability
|
Es wird auch empfohlen, aber nicht vorgeschrieben, Vorlagen und Formulare in das QMS aufzunehmen.
Zu den gängigen Beispielen für Vorlagen und Formulare, die nützlich sind, gehören:
Die ISO 13485 verlangt auch, dass die Organisation jede Anforderung, jedes Verfahren, jede Tätigkeit und jede Vereinbarung, die nach den geltenden gesetzlichen Bestimmungen dokumentiert werden müssen, einführt, umsetzt und aufrechterhält. Aus diesem Grund sind die oben aufgeführten 31 dokumentierten Verfahren möglicherweise nicht ausreichend, wenn das QMS den Anforderungen der MDR oder IVDR entsprechen soll.
Die ISO 13485 verlangt, dass die oberste Leitung einer Organisation eine Qualitätspolitik einführt und aufrechterhält. Die Qualitätspolitik sollte eine Erklärung sein, die die Verpflichtung der Organisation zur Erfüllung der Kunden- und gesetzlichen Anforderungen darlegt. Sie sollte auch die Verpflichtung der Organisation zur Aufrechterhaltung der Wirksamkeit ihres Qualitätsmanagementsystems (QMS) aufzeigen.
Häufig enthalten Qualitätsrichtlinien Informationen über die kontinuierliche Verbesserung des QMS. Dabei handelt es sich um eine Übernahme von Anforderungen aus der ISO 9000-Normenreihe, die das Konzept der kontinuierlichen Verbesserung anwendet. Da es sich bei der ISO 13485 um eine Regelungsnorm handelt, enthält sie keine Forderung nach kontinuierlicher Verbesserung, sondern nur nach Erreichen des geforderten Niveaus. Über das geforderte Niveau hinauszugehen, kann aus geschäftlicher Sicht wünschenswert sein, ist aber keine gesetzliche Anforderung der ISO 13485.
Die Anforderung, das QMS kontinuierlich zu verbessern, kann auch dann gelten, wenn die Organisation die Anforderungen der MDR erfüllen will, da Artikel 10, Abschnitt 9 der MDR dies vorschreibt:
Hersteller von Produkten, die keine Prüfprodukte sind, müssen […] ein Qualitätsmanagementsystem kontinuierlich verbessern.
Nachstehend finden Sie ein Beispiel für eine allgemeine Qualitätspolitik als Referenz.

Die Qualitätspolitik muss mit den allgemeinen Geschäftszielen der Organisation übereinstimmen und einen Rahmen für die Festlegung von Qualitätszielen bieten. Die Qualitätsziele sollten messbar sein und mit der Qualitätspolitik der Organisation übereinstimmen.
Beispiele für Qualitätsziele nach ISO 13485 könnten sein:
Bitte beachten Sie, dass Qualitätsziele manchmal auf Meilensteinen basieren und keine kontinuierliche Messung darstellen. Dies ist nützlich zu wissen, wenn man mit Qualitätszielen für Start-ups arbeitet, die keine kontinuierlichen Abläufe mit einer Vielzahl von Datenpunkten haben. Ein Beispiel für ein solches meilensteinbasiertes Qualitätsziel kann sein:
Der Begriff “wirksam” wird sowohl im Zusammenhang mit Anforderungen an die Qualitätspolitik als auch mit den Qualitätszielen verwendet. Die ISO 13485 bezieht sich oft auf “effektiv”, aber nie auf “effizient”. Die Worte “effektiv” und “effizient” beziehen sich beide auf das Erreichen eines Ergebnisses, jedoch mit einem wichtigen Unterschied.
Die “Effektivität” misst, inwieweit die geplanten Aktivitäten durchgeführt und die geplanten Ergebnisse erreicht werden, und die “Effizienz” fügt dem eine weitere Dimension hinzu, indem sie angibt, wie viele Ressourcen zur Erreichung des Ergebnisses aufgewendet wurden.
Aus rechtlicher Sicht ist es nie ein Erfordernis, effizient zu sein; es ist nur ein Erfordernis, effektiv zu sein. Aus geschäftlicher Sicht kann es jedoch wichtig sein, effizient zu sein.
Die oberste Leitung wird in der ISO 9000:2015 definiert als “eine Person oder Gruppe von Personen, die eine Organisation auf höchster Ebene leitet und kontrolliert”. Die ISO 13485 besagt, dass die oberste Leitung für die Einrichtung und Aufrechterhaltung des Qualitätsmanagementsystems (QMS) verantwortlich ist und sicherstellt, dass es die Anforderungen der Kunden und der Behörden wirksam erfüllt.
Die ISO 13485 verlangt von Organisationen, ein dokumentiertes Verfahren für die Managementbewertung einzuführen. Die Managementbewertung ist eine entscheidende Komponente des Qualitätsmanagementsystems, da sie der obersten Leitung die Möglichkeit bietet, die Leistung des Systems zu bewerten, verbesserungswürdige Bereiche zu ermitteln und Entscheidungen über die Ressourcenzuweisung zu treffen, um sicherzustellen, dass das QMS mit den strategischen Zielen der Organisation und den gesetzlichen Anforderungen übereinstimmt.

Eine der wichtigsten Komponenten des Qualitätsmanagements ist die Durchführung von Audits, um sicherzustellen, dass das QMS wirksam ist.
Im Großen und Ganzen gibt es drei verschiedene Kategorien von Prüfungen, jede mit ihrer eigenen Bedeutung und ihrem eigenen Zweck.
Erstparteien-Audits, auch bekannt als interne Audits, werden durchgeführt, um die fortdauernde Eignung, Angemessenheit und Wirksamkeit des Qualitätsmanagementsystems festzustellen und Verbesserungsmöglichkeiten zu ermitteln. Dies ist die Art von Audit, die von der ISO 13485 gefordert wird.
Second-Party-Audits oder externe Audits werden von externen Anbietern durchgeführt, um das Vertrauen in die Fähigkeiten eines externen Lieferanten zu gewinnen und zu erhalten. Diese Art von Audits wird von der ISO 13485 nicht immer explizit gefordert, aber es ist sehr üblich, Second-Party-Audits für die Lieferantenbewertung zu nutzen. Dies nicht zu tun, sollte als Ausnahme von der Regel betrachtet werden.
Schließlich gibt es zwei Arten von Audits durch Dritte: Zertifizierungs- und/oder Akkreditierungsaudits und gesetzlich vorgeschriebene oder regulatorische Audits. Der Zweck dieser Audits durch Dritte besteht darin, die geltenden gesetzlichen und behördlichen Anforderungen zu erfüllen. Zu dieser Kategorie gehören Audits durch benannte Stellen und die FDA.

Interne Audits eines Qualitätsmanagementsystems sollten in geplanten Abständen durchgeführt werden, um sicherzustellen, dass das System wirksam umgesetzt und aufrechterhalten wird. Die ISO 13485 enthält nicht viele Details zur Durchführung von Audits, die ISO 19011:2018 hingegen schon. Die aktuelle Version der Norm ISO 19011 finden Sie hier.
Zweck des internen Audits ist es, etwaige Nichtkonformitäten oder verbesserungswürdige Bereiche innerhalb des Qualitätsmanagementsystems zu ermitteln.
Der interne Auditprozess sollte einem dokumentierten Verfahren folgen und einen Zeitplan für die geplanten Audits, Kriterien für die Auswahl der Auditoren und Verfahren für die Durchführung des Audits umfassen.
Die ISO 13485 sagt nicht viel über den Zeitplan aus, aber der übergeordnete Zeitplan wird in der ISO 19011 als Auditprogramm bezeichnet.
Das Auditprogramm ist eine Reihe von einem oder mehreren Audits, die für einen bestimmten Zeitraum geplant und auf einen bestimmten Zweck ausgerichtet sind. Für jede Prüfung sollte ein Prüfungsplan erstellt werden. Ein Auditplan enthält eine Beschreibung der Aktivitäten und Vorkehrungen für ein Audit.
Der Begriff Auditplan wird oft fälschlicherweise als Bezeichnung für ein Auditprogramm verwendet. Die nachstehende Übersicht zeigt die Beziehung zwischen einem Auditprogramm und Auditplänen.

Das Audit muss von geschultem Personal durchgeführt werden, das unabhängig von dem Bereich ist, der auditiert wird. Der Auditor oder das Auditteam sollte relevante Unterlagen prüfen, Mitarbeiter befragen und Prozesse beobachten, um festzustellen, ob das Qualitätsmanagementsystem die Anforderungen der ISO 13485 und andere anwendbare Auditkriterien erfüllt.
Im Anschluss an die Prüfung sollten bei Bedarf Abhilfemaßnahmen ergriffen werden, ebenso wie Folgemaßnahmen und eine Überprüfung der Ergebnisse. Aufzeichnungen über alle durchgeführten Audits müssen aufbewahrt werden.
Die Norm ISO 13485 verlangt von den Organisationen, “einen risikobasierten Ansatz für die Kontrolle der für das Qualitätsmanagementsystem erforderlichen Prozesse anzuwenden“. In diesem Zusammenhang muss sowohl das Risiko in Bezug auf die Schädigung von Menschen, Eigentum und der Umwelt berücksichtigt werden, wie es in der ISO 14971 gefordert wird, als auch das Risiko der Nichteinhaltung von Vorschriften.
Darüber hinaus verlangt die ISO 13485, dass die Organisation einen oder mehrere Prozesse für das Risikomanagement bei der Produktrealisierung dokumentiert, wobei Aufzeichnungen über die im Rahmen des Risikomanagementprozesses durchgeführten Risikomanagementaktivitäten zu führen sind.
ISO 13485 verweist für weitere Informationen zum Risikomanagement auf ISO 14971. Diese Risikomanagementnorm ist mit der MDR und der IVDR harmonisiert und ist zusätzlich eine anerkannte Konsensnorm für den US-Markt.
Die Arbeit mit dem Risikomanagement in Bezug auf die Sicherheit des Medizinprodukts sollte in Übereinstimmung mit den Anforderungen der ISO 14971 durchgeführt werden. Dieser Risikomanagementprozess muss alle mit dem Produkt verbundenen Gefahren und Risiken während seines gesamten Lebenszyklus identifizieren, analysieren, bewerten und kontrollieren, von der ersten Konzeption und Auslegung bis hin zu Produktion, Vertrieb, Verwendung und Stilllegung.

Die ISO 13485 verlangt, dass Medizinprodukteunternehmen eine Infrastruktur einrichten und unterhalten, die für die Herstellung sicherer und wirksamer Medizinprodukte geeignet ist. Dazu gehören die physische Umgebung, wie Gebäude und Einrichtungen sowie die im Herstellungsprozess verwendeten Geräte und Werkzeuge.

Die Arbeitsumgebung muss so gestaltet sein, dass Verwechslungen und Kontaminationen vermieden werden und die Herstellung der Medizinprodukte in einer kontrollierten und sauberen Umgebung erfolgt. Dazu gehören Maßnahmen wie Luftfiltersysteme, Temperatur- und Feuchtigkeitskontrolle sowie die regelmäßige Reinigung und Wartung der Geräte.
Im Bereich der Infrastruktur und der Arbeitsumgebung muss eine Dokumentation geführt werden. Dazu gehören nicht nur die genauen Anforderungen an die Infrastruktur, sondern auch alle Aufzeichnungen über die Instandhaltung dieser Infrastruktur.
Außerdem müssen die Gesundheits-, Sauberkeits- und Bekleidungsvorschriften sorgfältig dokumentiert werden. Diese Faktoren spielen eine entscheidende Rolle bei der Aufrechterhaltung von Qualität und Sicherheit bei der Herstellung von Medizinprodukten.
Außerdem ist es wichtig, dass Mitarbeiter, die vorübergehend unter besonderen Umgebungsbedingungen arbeiten, wie z. B. in einem Reinraum, kompetent sind oder von einer kompetenten Person beaufsichtigt werden.
Schließlich ist eine strenge Kontrolle der Kontamination und Sauberkeit während der Produktionsphase von entscheidender Bedeutung. Dazu sagt die ISO 13485 gemäß 6.4.2:
Die Organisation muss die Anforderungen an die Kontrolle der Kontamination mit Mikroorganismen oder Partikeln dokumentieren und die erforderliche Sauberkeit während der Montage- oder Verpackungsprozesse aufrechterhalten.
Dies wird in der Regel durch die Produktion in Reinräumen erreicht. Reinräume werden wiederum von mehreren Normen abgedeckt, zum Beispiel in der Normenreihe ISO 14644.

ISO 13485 verlangt, dass die Organisation dokumentierte Anforderungen an das Produkt erstellt und aufrechterhält, einschließlich der geltenden gesetzlichen Anforderungen, der Anforderungen des Kunden, der Benutzerschulung und zusätzlicher Anforderungen.
Diese Anforderungen sollten während des gesamten Produktlebenszyklus überprüft und aktualisiert werden, um sicherzustellen, dass die Produktanforderungen definiert und dokumentiert sind, die gesetzlichen Anforderungen erfüllt werden und eine Benutzerschulung verfügbar ist, falls eine benötigt wird. Alle Änderungen sind zu dokumentieren, und Aufzeichnungen zu Überprüfungen und Maßnahmen sind aufzubewahren.
Die ISO 13485 verlangt von Medizinprodukteunternehmen die Einrichtung und Aufrechterhaltung eines Systems zur Identifizierung, Dokumentation, Überprüfung, Kontrolle und Sicherung von Kundeneigentum. Dazu gehört jegliches Eigentum, das vom Kunden oder einem externen Anbieter zur Verwendung bei der Herstellung oder Wartung von Medizinprodukten bereitgestellt wird.
Es ist ein weit verbreiteter Irrglaube, dass Kundeneigentum nur materielle Güter umfasst. Auch geistiges Eigentum und personenbezogene Daten können als Kundeneigentum betrachtet werden. Siehe GDPR und HIPAA für weitere Informationen zu diesem Bereich.
Beispiele für Kundeneigentum sind:
Eine wirksame Kommunikation mit den Kunden ist ein wichtiger Aspekt der ISO 13485. Die Norm schreibt vor, dass Medizinprodukteunternehmen Kommunikationsprozesse mit ihren Kunden einrichten und aufrechterhalten müssen, um deren Bedürfnisse und Erwartungen zu erfüllen.
Dazu gehört die Bereitstellung von Informationen über das Produkt, seinen Verwendungszweck und alle damit verbundenen Einschränkungen oder Risiken. Dazu gehört auch, Beschwerden zu dokumentieren und zu untersuchen, Abhilfemaßnahmen zu ergreifen und Aufzeichnungen über alle Kundenbeschwerden und Rückmeldungen zu führen.
Die Kommunikation mit den Aufsichtsbehörden ist ein wesentlicher Bestandteil der Einhaltung der ISO 13485. Die Norm verlangt, dass Medizinprodukteunternehmen mit den Aufsichtsbehörden in Übereinstimmung mit den geltenden Vorschriften kommunizieren, um sicherzustellen, dass sie über alle Änderungen der Vorschriften oder Anforderungen, die ihre Produkte betreffen könnten, informiert sind.
Die ISO 13485 verlangt von Medizinprodukteunternehmen, dass sie einen dokumentierten Entwurfs- und Entwicklungsprozess einrichten und aufrechterhalten.
Dieser Prozess muss für den spezifischen Gerätetyp geeignet sein und umfasst eine Reihe von Aktivitäten, die mit dem ersten Konzept beginnen und bis zur Freigabe des Endprodukts für den Vertrieb andauern.
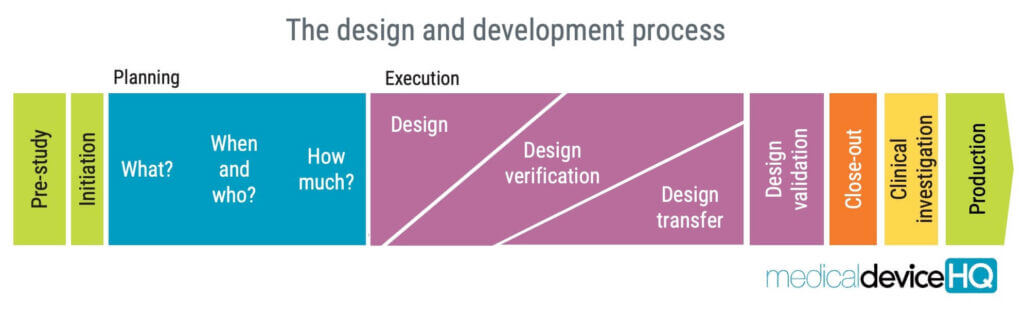
Die Anforderungen an Design und Entwicklung in ISO 13485 zielen darauf ab, sicherzustellen, dass das Medizinprodukt den vorgesehenen Verwendungszweck erfüllt, sicher und wirksam ist und den gesetzlichen Anforderungen entspricht. Aus der Sicht des Herstellers kann der Prozess auch dazu genutzt werden, ein Produkt zu entwickeln, mit dem die Kunden zufrieden sind.
Der Entwurfs- und Entwicklungsprozess umfasst mehrere Bereiche, wie Entwurfs- und Entwicklungsplanung, Entwurfs- und Entwicklungsinputs, Entwurfs- und Entwicklungsoutputs, Entwurfs- und Entwicklungsüberprüfung, Entwurfs- und Entwicklungsverifizierung, Entwurfs- und Entwicklungstransfer und Entwurfs- und Entwicklungsvalidierung.
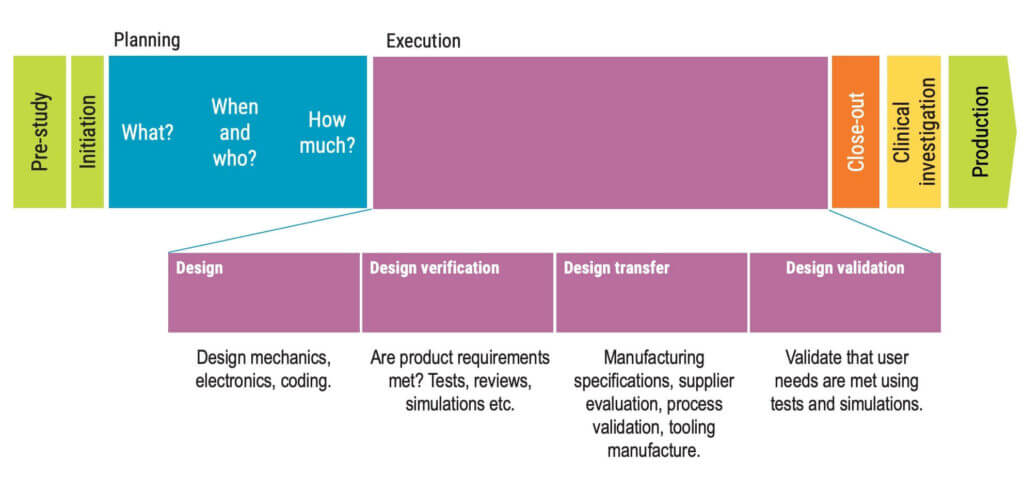
Jede Stufe hat spezifische Anforderungen, die von der Organisation festgelegt werden sollten, um die Qualität des Medizinprodukts zu gewährleisten.
Obwohl eine gezielte Gestaltung der Gebrauchstauglichkeit in der ISO 13485 nur ein paar Mal erwähnt wird, ist sie eine entscheidende Komponente des Risikomanagements, die von der FDA für den US-Markt und von der MDR und IVDR für den EU-Markt gefordert wird.
Die Norm ISO 13485 verlangt von Unternehmen, die mit der Verwendung eines Produkts verbundenen Risiken zu ermitteln und zu bewerten, einschließlich der Risiken im Zusammenhang mit der Gebrauchstauglichkeit. Die Prüfung der Gebrauchstauglichkeit ist ein wichtiges Instrument zur Identifizierung und Minderung dieser Risiken.
Durch das Testen des Geräts mit repräsentativen Anwendern in realistischen Nutzungsszenarien können Organisationen potenzielle Probleme bei der Benutzerfreundlichkeit erkennen und Designänderungen vornehmen, um die Sicherheit und Wirksamkeit des Geräts zu verbessern.
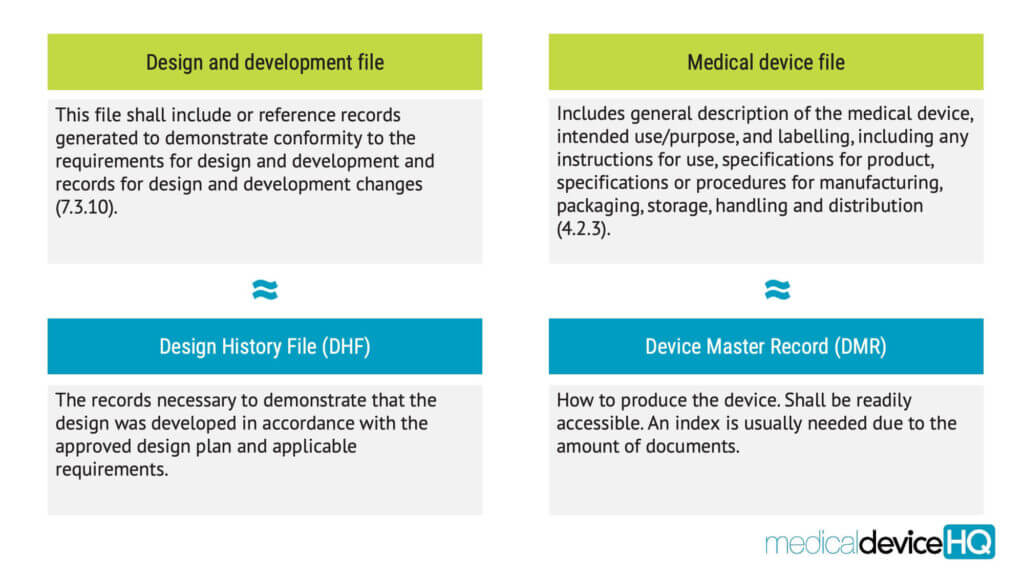
Die Produktdokumentation sollte während des gesamten Produktlebenszyklus geführt werden, von der Konzeption und Entwicklung über die Produktion und den Vertrieb bis hin zur Überwachung nach dem Inverkehrbringen.
Die Organisationen sollten auch Verfahren für die Aufbewahrung der Produktdokumentation festlegen, einschließlich einer Entwicklungsakte und einer Medizinprodukteakte. Im QSR werden diese als Design History File (DHF) und Device Master Record (DMR) bezeichnet.
Die MDR und die IVDR werden sich in den Anhängen II und III auf die “technische Dokumentation” beziehen, was sich mit den Begriffen der ISO 13485 überschneidet.
Die Lieferantenbewertung ist ein wichtiger Aspekt der ISO 13485 und umfasst die Beurteilung der Fähigkeiten und Leistungen der Lieferanten, um sicherzustellen, dass sie die Anforderungen der Organisation erfüllen. Dazu kann gehören, dass die Lieferanten auch die Anforderungen der ISO 13485 erfüllen sollten.
Der Bewertungsprozess umfasst:
Die Kriterien für die Bewertung müssen Faktoren wie die Fähigkeit, die Anforderungen der Organisation zu erfüllen, und die Leistung des Lieferanten umfassen. Aus regulatorischer Sicht ist der Preis in der Regel nicht Teil der Kriterien. Die Bewertung kann durch den Erhalt von Prototypen, Lieferantenbefragungen, Audits oder Besuche vor Ort erfolgen.
Es ist üblich, dass Medizinprodukteunternehmen eine Liste zugelassener Lieferanten erstellen, aber es ist nicht ausdrücklich vorgeschrieben, eine solche Liste zu führen, da es andere Möglichkeiten gibt, um zu verfolgen, welche Lieferanten zugelassen sind und welche nicht.
Organisationen müssen über einen Plan zur Überwachung und Neubewertung von Lieferanten verfügen. Wenn ein Lieferant ein Produkt liefert, das nicht der Spezifikation entspricht, wird dies gemäß ISO 13485, 8.3 als Nichtkonformität betrachtet.
Es ist erforderlich, genaue Aufzeichnungen über Lieferanten- und Einkaufsinformationen zu führen. Dies hilft den Unternehmen auch bei der Bewertung der Lieferantenleistung. Durch die Verfolgung der Leistungskennzahlen von Lieferanten, wie z. B. pünktliche Lieferung, Produktqualität und Reaktionsfähigkeit bei Problemen, können Unternehmen fundierte Entscheidungen darüber treffen, mit welchen Lieferanten sie weiterhin zusammenarbeiten und welche sie ersetzen sollten.
Die ISO 13485 verlangt, dass Medizinprodukteunternehmen Verfahren einführen und aufrechterhalten, um sicherzustellen, dass die eingekauften Produkte mit den festgelegten Einkaufsinformationen übereinstimmen. Diese Verfahren sollten Anforderungen für die Genehmigung von eingekauften Produkten oder Dienstleistungen sowie für die Kontrolle von nicht konformen Produkten oder Dienstleistungen enthalten.
Die ISO 13485 verlangt außerdem, dass Medizinprodukteunternehmen Aufzeichnungen über ihre Einkaufsaktivitäten führen, um die Anforderungen an die Rückverfolgbarkeit zu erfüllen.
Gekaufte Waren und verarbeitete Materialien, die für die Herstellung von Medizinprodukten erworben werden, müssen aufbewahrt werden, um sicherzustellen, dass die daraus entstehenden Produkte den Anforderungen entsprechen. Dies kann Verfahren für die Lagerung und den Schutz der Waren, Materialien und schließlich der fertigen Produkte durch eine geeignete Verpackung umfassen. Die Verpackung sollte immer als integraler Bestandteil des Designs betrachtet werden.
Medizinprodukte, die steril an den Kunden geliefert werden, müssen in einem Sterilbarrieresystem verpackt sein und über eine Schutzverpackung verfügen, die so konzipiert ist, dass eine Beschädigung des Sterilbarrieresystems und seines Inhalts von der Montage bis zur Verwendung verhindert wird. Das Sterilbarrieresystem muss unter dem Gesichtspunkt der Gebrauchstauglichkeit gemäß ISO 11607-1:2020 bewertet werden. Die Akzeptanzkriterien für eine solche Bewertung sind jedoch in der ISO 11607-1 nicht definiert.
Zugekaufte Produkte müssen einer Eingangs- oder Wareneingangskontrolle unterzogen werden. In der ISO 13485 wird dies als Verifizierung der eingekauften Produkte bezeichnet. Es wird nicht ausdrücklich erwähnt, dass bei der Überprüfung der eingekauften Produkte statistische Methoden angewandt werden sollten, aber es wird mit Sicherheit erwartet, dass Stichprobenpläne auf der Grundlage statistischer Überlegungen verwendet werden oder dass 100 % der eingekauften Produkte überprüft werden. Alle Produkte, die sich als nicht konform erweisen, werden gemäß den Verfahren von behandelt.
Während des gesamten Produktionsprozesses müssen die in der Produktion verwendeten Waren und die daraus hergestellten Produkte identifizierbar sein. Diese Anforderungen finden sich in Abschnitt 7.5.8 der ISO 13485.
In der Praxis kann die Kennzeichnung beispielsweise dadurch erfolgen, dass die Waren in etikettierte Kartons gelegt oder mit Aufklebern versehen werden. Gegebenenfalls muss auch ein System zur Zuordnung von UDI zu den Medizinprodukten dokumentiert werden.
Während des gesamten Produktionsprozesses müssen geeignete Kontrollen durchgeführt werden, um sicherzustellen, dass die Produkte den Anforderungen entsprechen. Die Überwachung muss entsprechend den Plänen erfolgen. Wenn Messungen durchgeführt werden, müssen die Messgeräte kalibriert werden. Denken Sie daran, dass prozessbegleitende Kontrollen in der Produktion häufig Maßnahmen zur Risikokontrolle sind; Schutzmaßnahmen im Herstellungsprozess gemäß der Norm ISO 14971 zum Risikomanagement.
Für jedes Medizinprodukt oder jede Charge von Medizinprodukten, die hergestellt werden, muss ein Chargenprotokoll erstellt werden. Ein gebräuchlicher Begriff für Chargenprotokoll ist Device History Record, oder kurz DHR aus der Qualitätssystemverordnung.
Die Produktfreigabe, d. h. die Erlaubnis, das Produkt in Verkehr zu bringen, muss nach dokumentierten Vereinbarungen erfolgen. Dies bedeutet in der Regel, dass dokumentiert werden sollte, welche Tests, Aufzeichnungen, Maßnahmen, Überprüfungen und Genehmigungen von Produktionsaufzeichnungen vor der Freigabe des Produkts abgeschlossen sein müssen. Für den EU-Markt wird die Produktfreigabe von der PRRC (Person Responsible for Regulatory Compliance) durchgeführt.
Wenn das Medizinprodukt installiert werden muss, müssen die Anforderungen an die Installation dokumentiert werden.
Die ISO 13485 enthält an sich keine Einzelheiten zu den Anforderungen an die Rückverfolgbarkeit, außer dass Sie Verfahren für die Rückverfolgbarkeit festlegen müssen. Für weitere Einzelheiten verweist die ISO 13485 auf die geltenden gesetzlichen Anforderungen an die Rückverfolgbarkeit.
Als allgemeines Minimum müssen der Hersteller und seine Vertriebshändler verfolgen, an wen und wohin die Medizinprodukte geliefert werden. Dies muss geschehen, um sicherzustellen, dass im Bedarfsfall geeignete Sicherheitskorrekturmaßnahmen vor Ort durchgeführt werden können.
Detailliertere Anforderungen an die Rückverfolgbarkeit gelten für implantierbare Produkte gemäß 7.5.9.2.
Die Hersteller können sich aus Gründen des Risikos für die Patienten und des Geschäftsrisikos für eine detailliertere Rückverfolgbarkeit entscheiden. Zu wenig Informationen über die an die Kunden gelieferten Medizinprodukte können dazu führen, dass jedes auf dem Markt befindliche Medizinprodukt zurückgerufen werden muss, anstatt eine bestimmte Charge oder ein einzelnes Produkt.
Die Anforderungen der ISO 13485 an die Prozessvalidierung sind ein wesentlicher Bestandteil zur Gewährleistung der Sicherheit und Wirksamkeit von Medizinprodukten.
Die Prozessvalidierung ist der Prozess, bei dem der Nachweis erbracht wird, dass ein Prozess durchgängig ein Produkt erzeugt, das die vorgegebenen Spezifikationen und Qualitätsmerkmale erfüllt. Dies wird durch eine Reihe von Aktivitäten erreicht, die die Planung, den Entwurf, die Qualifizierung und die laufende Überwachung des Prozesses umfassen.

Beispiele für Herstellungsprozesse von Medizinprodukten, die eine Prozessvalidierung erfordern, sind Spritzgießen, Drucken, Kleben, Löten, Schweißen und Wärmebehandlung.
Weitere Beispiele sind der Sterilisationsprozess, der Montageprozess und der Verpackungsprozess. So muss beispielsweise der Sterilisationsprozess validiert werden, um sicherzustellen, dass er Mikroorganismen auf dem Produkt wirksam abtötet, ohne dessen Funktionalität zu beeinträchtigen oder nachteilige Auswirkungen auf den Patienten zu haben.
ISO 13485 verlangt auch, dass Software, die im QMS, in der Produktion und im Service sowie zur Überwachung und Messung von Anforderungen eingesetzt wird, validiert werden muss. Das heißt, wenn die Organisation ein eQMS einsetzt, muss dieses validiert werden.

Lesen Sie mehr über Prozessvalidierung in Bezug auf Produktionsprozesse im Dokument der Global Harmonization Task Force und über die Validierung von Software, die im QMS verwendet wird, in ISO/TR 80002-2:2017 Medical device software – Part 2: Validation of software for medical device quality systems.
Die Anforderungen der ISO 13485 an nicht konforme Produkte sind ein wesentlicher Aspekt der Norm. Nichtkonforme Produkte sind Produkte, die nicht den festgelegten Anforderungen entsprechen.
Die Norm verlangt, dass Organisationen dokumentierte Verfahren zur Identifizierung, Dokumentation, Bewertung, Aussonderung und Entsorgung fehlerhafter Produkte einführen und aufrechterhalten.
Diese Verfahren sollten die Identifizierung, Dokumentation, Aussonderung, Bewertung und Beseitigung fehlerhafter Produkte sowie die Durchführung von Korrekturmaßnahmen umfassen, um ein erneutes Auftreten des Fehlers zu verhindern.
Die Norm verlangt auch, dass die Organisation fehlerhafte Produkte überprüft und analysiert, um Trends zu erkennen und geeignete Maßnahmen zu ergreifen.
Wenn ein fehlerhaftes Produkt vor der Auslieferung entdeckt wird, ist es wichtig, Maßnahmen zu ergreifen, um zu verhindern, dass das Produkt an den Kunden ausgeliefert wird.
Das fehlerhafte Produkt sollte ausgesondert und deutlich gekennzeichnet werden, um zu verhindern, dass es versandt oder verwendet wird. Nicht konforme Produkte werden häufig mit Aufklebern gekennzeichnet und/oder in einem separaten Bereich untergebracht.
Abhängig von der Kritikalität und dem Risiko der Nichtkonformität, kann das Produkt:
Wird eine Nacharbeit durchgeführt, muss die Organisation überprüfen, ob das Produkt noch den Anforderungen und der vorgesehenen Verwendung entspricht. Eine häufige Frage ist, ob ein steriles Medizinprodukt erneut sterilisiert und dann auf den Markt gebracht werden kann. Einige Hersteller führen vorsorglich eine Validierung eines Re-Sterilisationsprozesses als Projektrisikokontrollmaßnahme durch, um nachzuweisen, dass das Produkt auch nach der Re-Sterilisation die Anforderungen erfüllt.
Über etwaige Nachbesserungen oder Zugeständnisse sind stets Aufzeichnungen zu führen.
Wird ein fehlerhaftes Produkt entdeckt, nachdem es an den Kunden ausgeliefert wurde, müssen je nach den Auswirkungen des Fehlers Maßnahmen ergriffen werden. Dies kann Folgendes umfassen:
Die Organisation muss über ein dokumentiertes Verfahren für die Herausgabe von Hinweisen im Falle eines solchen Ereignisses verfügen, das jederzeit einsatzbereit sein muss. Daraus erfolgt auch, dass im Falle von zeitweisen Betriebsschließungen (z.B. Betriebsurlaub) Mechanismen etabliert sein müssen, die sicherstellen, dass die notwendigen Schritte trotzdem ausgeführt werden können.
CAPA steht für Corrective Action and Preventive Action (Korrektur- und Vorbeugemaßnahmen) und ist ein wesentlicher Bestandteil des Qualitätsmanagementsystems nach ISO 13485. Der Zweck von CAPA ist es, zu verhindern, dass sich Nichtkonformitäten wiederholen oder auftreten. CAPA gilt sowohl für Nichtkonformitäten im Zusammenhang mit Medizinprodukten als auch für Dienstleistungen und das Qualitätsmanagementsystem.
Korrekturmaßnahmen sind definiert als Maßnahmen zur Beseitigung der Ursache einer Nichtkonformität und zur Verhinderung des erneuten Auftretens einer solchen. Korrekturmaßnahmen sind nicht mit Korrekturen zu verwechseln. Der Unterschied besteht darin, dass eine Korrektur eine einmalige Lösung für das vorliegende Problem darstellt, während Korrekturmaßnahmen verhindern, dass dasselbe Problem erneut auftritt.
Der erste Schritt bei Korrekturmaßnahmen besteht immer darin, zu untersuchen, warum die Nichtkonformität aufgetreten ist. Auf der Grundlage der Ergebnisse dieser Untersuchung müssen dann Maßnahmen ergriffen werden. Diese Untersuchung wird oft als Ursachenanalyse bezeichnet, obwohl dieser Begriff in der ISO 13485 nicht erwähnt wird. In der ISO 13485 heißt es lediglich “Ermittlung der Ursachen von Nichtkonformitäten”, was jedoch in den meisten Fällen als Ursachenanalyse verstanden werden sollte.
Wenn eine festgestellte Nichtkonformität ein geringes Risiko darstellt, kann es ausreichen, das Problem durch eine kleine Korrektur zu beheben. Ist das Risiko jedoch höher, verlangt die ISO 13485, dass Sie Korrekturmaßnahmen ergreifen, die in einem angemessenen Verhältnis zu den Auswirkungen der aufgetretenen Nichtkonformitäten stehen.

Ihr CAPA-Verfahren muss beinhalten, wie die Ursachen von Nichtkonformitäten ermittelt werden, was am häufigsten durch eine Ursachenanalyse geschieht. Eine Möglichkeit, dies zu tun, besteht darin, die Frage “Warum?” fünfmal zu stellen. Sie könnten auch eine qualitative Analyse durchführen oder ein Fischgrätendiagramm verwenden, um die Ursache zu ermitteln.
Sobald Sie die Grundursache ermittelt haben, sollten die Korrekturmaßnahmen geplant und dokumentiert werden, bevor die Umsetzung beginnt.
Nach der Umsetzung müssen Sie überprüfen, ob die Maßnahmen, die Sie ergreifen, auch Wirkung zeigen. Dazu müssen Sie prüfen, ob die getroffenen Maßnahmen:
Sie müssen auch die Wirksamkeit der Abhilfemaßnahmen überprüfen, um sicherzustellen, dass sie die von Ihnen geplanten Ergebnisse erzielen.
Vorbeugende Maßnahmen ähneln den korrigierenden Maßnahmen, mit dem Unterschied, dass vorbeugende Maßnahmen ohne vorangegangene Nichtkonformität eingeleitet werden.
Bei Präventivmaßnahmen geht es darum, das Auftreten potenzieller Nichtkonformitäten proaktiv zu verhindern, bevor etwas schief geht. Vorbeugende Maßnahmen werden in der Regel als Ergebnis von Entscheidungen eingeleitet, die im Rahmen einer Managementbewertung getroffen werden.

Gemäß ISO 13485:2016, 3.4, sind Beschwerden:
Schriftliche, elektronische oder mündliche Mitteilung, in der Mängel im Zusammenhang mit der Identität, Qualität, Haltbarkeit, Zuverlässigkeit, Gebrauchstauglichkeit, Sicherheit oder Leistung eines Medizinprodukts behauptet werden, das der Kontrolle der Organisation entzogen wurde, oder die sich auf eine Dienstleistung bezieht, die die Leistung solcher Medizinprodukte beeinträchtigt.
Jeder, der mit externen Personen in Kontakt kommt, die nicht zu dem Medizinprodukteunternehmen gehören, muss wissen, wenn er eine Beschwerde erhält. Dazu gehören zum Beispiel alle Personen, die möglicherweise in der Organisation ans Telefon gehen oder sich mit Kunden treffen könnten.
Es sei darauf hingewiesen, dass der QSR eine etwas andere Definition des Begriffs “Beschwerde” verwendet.
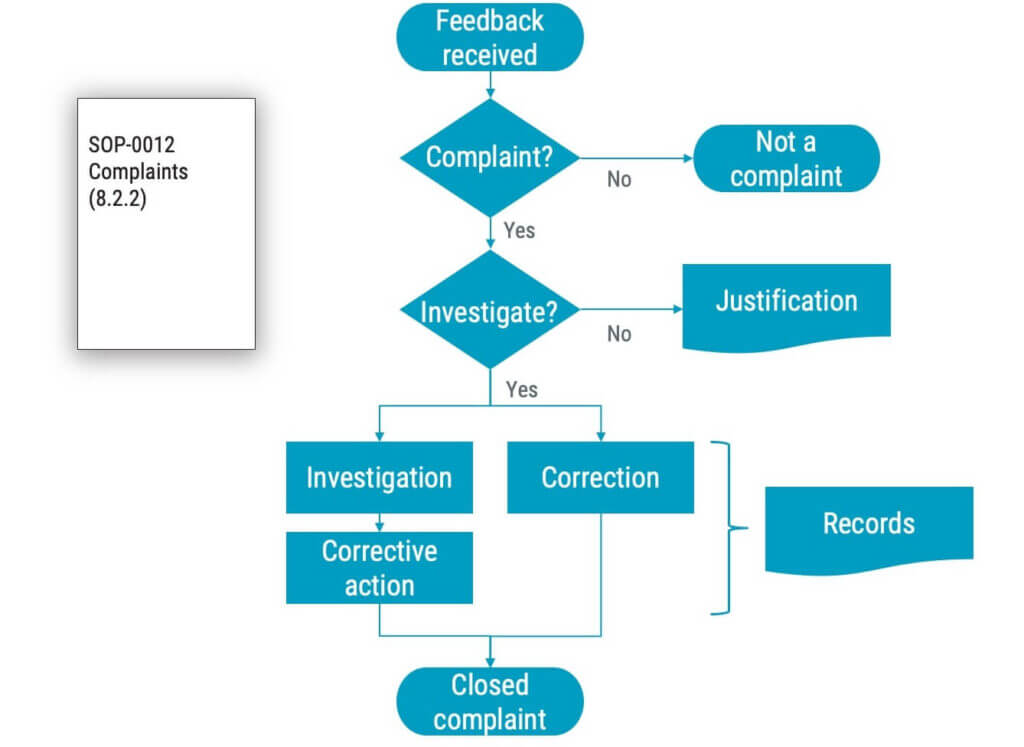
Gemäß ISO 13485 müssen Medizinprodukteunternehmen ein dokumentiertes Verfahren für die Entgegennahme, Bewertung und Untersuchung von Beschwerden einrichten. Darüber hinaus muss eine Beschwerde unter Umständen den Aufsichtsbehörden gemeldet werden, wenn sie eine Schädigung von Menschen oder eine mögliche Schädigung von Menschen beinhaltet. Aufzeichnungen über Beschwerden müssen aufbewahrt werden. Weitere Einzelheiten zum Umgang mit Beschwerden finden Sie in Abschnitt 8.2.2 der ISO 13485.
Ein Verständnis des Qualitätsmanagements für Medizinprodukte und der ISO 13485 ist für jeden, der in der Medizinprodukteindustrie tätig ist, von großer Bedeutung.
Qualitätsmanagement und die Implementierung eines effektiven und effizienten QMS sind entscheidende Erfolgsfaktoren für Unternehmen, die mit Medizinprodukten arbeiten. Es handelt sich um systematische Prozesse, die sicherstellen sollen, dass ein Produkt durchgängig die festgelegten Anforderungen erfüllt.
|
4.1.6 Validation of the application of computer software used in the quality management system
|
7.5.11 Preserving conformity of product to requirements during processing, storage, handling, and distribution
|
|
4.2.4 Controls needed to review, approve documents, update and re-approve documents
|
7.6 Procedures to ensure monitoring and measurement is done according to requirements
|
|
4.2.5 Controls needed for the identification, storage, security and integrity, retrieval, retention time and disposition of records
|
7.6 Validation of computer software used for monitoring and measurement
|
|
5.6.1 Management review
|
7.6 Calibration and verification
|
|
6.4.1 Monitor and control work environment if it has effect on product quality
|
8.2.1 Feedback
|
|
7.3.1 Design and development
|
8.2.2 Complaints handling
|
|
7.3.8 Transfer of design and development outputs to manufacturing
|
8.2.3 Notification of adverse events or issuance of advisory notices
|
|
7.3.9 Control of design and development changes
|
8.2.4 Responsibilities and requirements for planning and conduct of audits
|
|
7.4.1 Ensuring purchased product conforms to purchasing information
|
8.2.6 Monitoring and measuring characteristics of the product
|
|
7.5.1 Procedures for control of production
|
8.3.1 Responsibilities and authorities for identification, documentation, segregation, evaluation, and disposition of nonconforming product
|
|
7.5.6 Validation of processes
|
8.3.3 Issuing advisory notices
|
|
7.5.6 Validation of computer software used in production and service
|
8.3.4 Rework
|
|
7.5.7 Validation of processes for sterilization and sterile barrier systems
|
8.4 Collect and analyze appropriate data
|
|
7.5.8 Identification of production by suitable means through product realization
|
8.5.2 Reviewing nonconformities
|
|
7.5.8 Returned medical devices
|
8.5.3 Determining nonconformities and their causes
|
|
7.5.9.1 Traceability
|
Receive FREE templates and quarterly updates on upcoming courses that can help you in your career! Subscribe to our newsletter now.
When you submit this form, you will be sending personal information to medicaldevicehq.com. To comply with GDPR requirements, we need your consent to store and use the personal data you submit. Take a look at our Privacy policy for more details.
Special launch offer: 349 299 EUR for the online plan & 449 349 EUR for the online lifetime plan.