
As you may know, in December 2019, the new updated version of the ISO 14971 was released. What does this update mean to you? Use this checklist template if you need help going over your own procedures and documents for the update.
This video is an extract from the online course Risk Management for Medical Devices and ISO 14971:2019.
A systematic review vote in 2016 was taken by the ISO organisation and IEC on updating the ISO 14971:2007. The result of the vote was to reaffirm the standard, but there were comments on how it could be improved. The technical committee was assigned to update the standard and the technical report, which is a guidance to the implementation of ISO 14971.
But, the technical committee was also to maintain key concepts and the core approach to risk management. Basically, we, who have been working with the standard, have not been allowed to change the process.
You will find that it’s changed very little; risk management planning has been added on the side, and the names of the risk management report and production and post-production information have been updated. The updated names are two very welcome changes because the previous version of the standard indicated that this was just a report and information, whereas in fact, they were and still are two processes.

With the new ISO 14971:2019, you can say that the standard is better aligned with the general safety and performance requirements of the MDR and IVDR. It would, however, be more accurate to say that the MDR and IVDR are now better aligned with the process that has been described in the standard the whole time.
I have listed the 6 most important changes to the standard. I have ranked them in order of importance, based on my subjective opinion. And to be accurate, two of the things I will be bringing up are actually not changes in the standard, but they will still have an impact on how you work.
So, what you will be seeing first are the things that will have the biggest impact on a) your ways of working, b) your procedures, and c) records from risk.
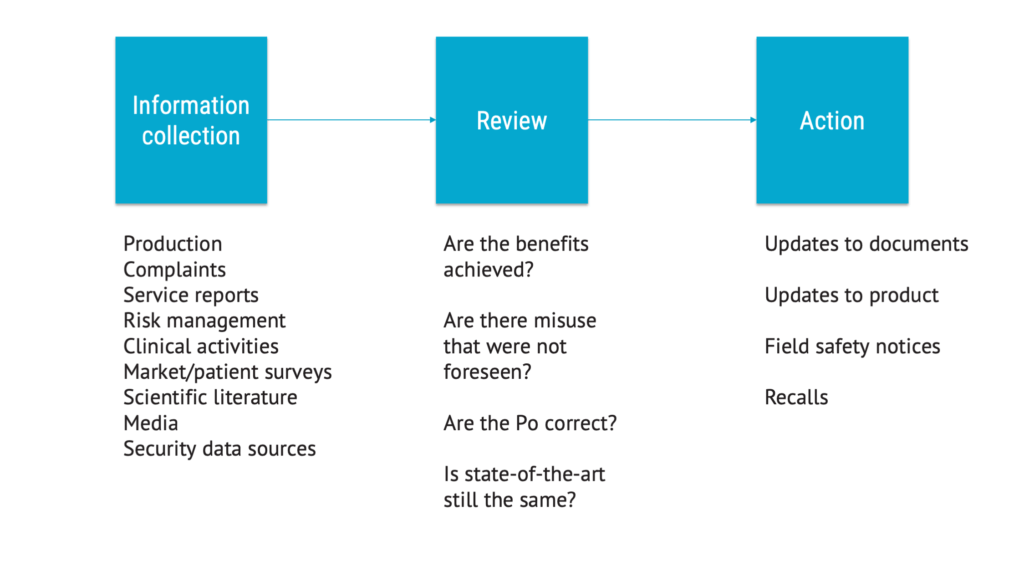
The chapter on production and post-production activities, formerly known as production and post-production information, is now much more comprehensive and granular. It describes a process that has more steps than before.
This makes perfect sense. This is about setting up an effective way of collecting information that can be used to maintain the risk management file. It is also much better aligned with current expectations on post-market surveillance.
People have been quite confused with regards to what inherent safety by design is. Many have believed that it only relates to the design, and that you cannot apply the same risk control option to manufacturing. There has been even more confusion as to what information for safety and disclosure of residual risk is. The wording used for these risk control measures have been updated in the standard.
On the left-hand side, you can see the risk control options in the old standard. The first one being inherent safety by design. On the right-hand side, you can see that the first risk control option refers to inherently safe design and manufacture. Hopefully, this will reduce the risk of people believing that the first risk control option can only be applied to the design.

Please, also note the differences in the third risk control option, where the old standard refers only to information for safety, and the new standard also mentions training to users.
Basically, only one word has been removed in the new definition, but it makes a big difference.
As you can see in this comparison, the word physical in the old definition of harm has been removed. The rest of the definition is identical. But, what this means is that risk management covers more risks than before, for example breach of personal integrity or stress when receiving an incorrect diagnosis.

You need to consider cyber security. This will only apply if your product contains software, or is a software in itself, or could have an impact on software. Not every security failure will lead to harm but they could, and that is why you need to include hazards relating to security breaches in your risk analysis. If you have software in your product, take a look at the risk analysis to make sure that you have considered:
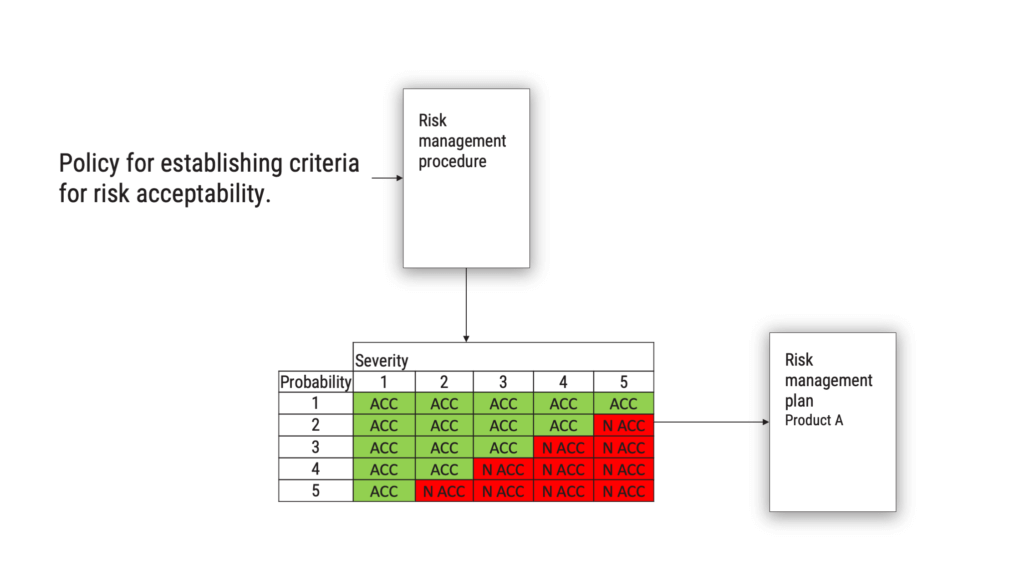
The requirement on defining and documenting a policy for establishing criteria for risk acceptability has been clarified in ISO/ TR 24971, there is no longer any excuse not to do this properly. If you ask a medical device manufacturer for a risk management policy, from 2007 and even today, they are likely to show you the risk evaluation matrix. This is not the intent of the standard.

What you should be doing, is to define and document a policy which is the starting point for your risk evaluation matrix or the criteria you use to determine if a risk is acceptable or not. The policy will go into your risk management procedure, and when you have applied your policy, the output will be the criteria for acceptability of risk. This should be found in your risk management plan.
What should the policy say? It could, for example, say that criteria shall be based on the generally acknowledged state-of-the-art, as determined from similar medical devices available on the market.
And lastly, the removed content deviations. In the EN ISO 4971 2012 version of the risk management standard, there were three Annex Z that described how the standard meets or does not meet the requirements of the three medical device directives. With the new MDR, and the new version of the standard, there are no longer any content deviations.
However, there is one thing mentioned in Annex Z of the new standard, and this related to the previous topic I brought up about policy for risk acceptability. You should comply with requirements for sales to the EU and the MDR, the policy for establishing criteria for risk acceptability and the resulting criteria must take general safety and performance requirements into account, meaning for example that risks must be reduced as far as possible as long as it does not adversely affect the benefit risk ratio.
Do you need to transition and update your procedures, templates and risk management documents and records to ISO 14971:2019? If so, download the ISO 14971 transition checklist.
Get instant access to our online Risk Management for Medical Devices and ISO 14971:2019 course right here. In 10 hours, you can learn more about how to develop new medical devices and maintain them in organisations where design control requirements apply. This course is taken by quality assurance, project management, design engineering or those involved in R&D and product development teams.

Peter Sebelius is a highly esteemed trainer, consultant and entrepreneur in the medical device industry. He is a member of the Joint Working Group that is revising the ISO 13485 and ISO 14971 standards.
He has vast ‘hands on’ experience, having developed, amongst other things, a mechanical chest compression device and an ex vivo perfusion machine for lungs. He has received numerous awards including the Great Design Award and the title “This year’s specialist” by Veckans affärer.
Receive FREE templates and quarterly updates on upcoming courses that can help you in your career! Subscribe to our newsletter now.
When you submit this form, you will be sending personal information to medicaldevicehq.com. To comply with GDPR requirements, we need your consent to store and use the personal data you submit. Take a look at our Privacy policy for more details.


